
中山大学陈样新教授团队在《Hypertension》发表最新研究:揭示内皮细胞SMAD4缺失促进肺高压发病的关键机制
2025-04-16 刘少飞 MedSci原创

该研究首次从分子与结构层面揭示了内皮SMAD4缺失如何通过影响细胞连接与基质稳态,驱动肺动脉高压的发生。
肺高压(Pulmonary Hypertension, PH)作为一种进展性肺血管疾病,严重影响患者的生活质量及生存率,目前仍缺乏根治性的治疗手段。近日,中山大学孙逸仙纪念医院及中山大学眼科中心陈样新教授、张峰教授、王景峰教授团队在《Hypertension》期刊上发表题为**“Endothelial SMAD4 Deficiency Promotes Pulmonary Hypertension by Impairing Cell Adhesion and Extracellular Matrix Organization”的研究论文,系统阐明了内皮细胞中SMAD4缺失如何通过破坏细胞黏附与细胞外基质结构,促进肺高压的发生和进展**,为靶向干预PH提供了全新视角与潜在策略。
肺高压的复杂病因与信号通路异常
肺高压的发病机制复杂,涉及肺小动脉重构、内膜增生、平滑肌细胞(SMC)异常增殖以及炎症反应。其中,BMPR2(骨形态发生蛋白受体2)信号通路异常已被证实在多种遗传性和散发性肺动脉高压中发挥关键作用。BMPR2的突变会扰乱TGF-β/BMP家族相关信号,导致内皮细胞功能障碍与血管重塑加剧。
SMAD4作为TGF-β/BMP通路的核心转录因子,其在血管发育与稳态中的作用已有大量研究,但其在肺高压中的细胞特异性功能和机制尚未完全明确。本研究首次聚焦于内皮细胞特异性SMAD4缺失在肺高压中的致病作用,填补了该领域的研究空白。
研究亮点一:内皮细胞SMAD4缺失而非SMC缺失是PH发病关键
研究团队通过构建不同类型的组织特异性基因敲除小鼠模型,发现:
- 全身性SMAD4缺失小鼠自发出现肺高压表型,并在低氧条件下加重;
- 平滑肌细胞中敲除SMAD4并不会诱导肺高压发生;
- 而在内皮细胞中特异性敲除SMAD4,则显著诱发肺高压,并表现为肺血管重构、右心负荷增加等典型病理改变。
这一结果明确了肺高压的发病更依赖于内皮SMAD4信号的完整性,提示靶向修复内皮功能或将成为更有效的治疗方向。
研究亮点二:揭示细胞黏附与ECM破坏为关键机制
进一步的体内外实验显示,内皮细胞SMAD4缺失不仅诱导细胞衰老和凋亡,还导致:
- 内皮细胞间连接结构紊乱;
- 细胞-基质黏附功能明显减弱;
- 基底膜与细胞外基质(ECM)稳定性受损;
- 血管通透性异常增强,促发炎性细胞浸润。
这些改变共同破坏了肺血管屏障功能,形成炎性微环境与重塑信号持续激活的恶性循环,从而推动PH的持续进展。
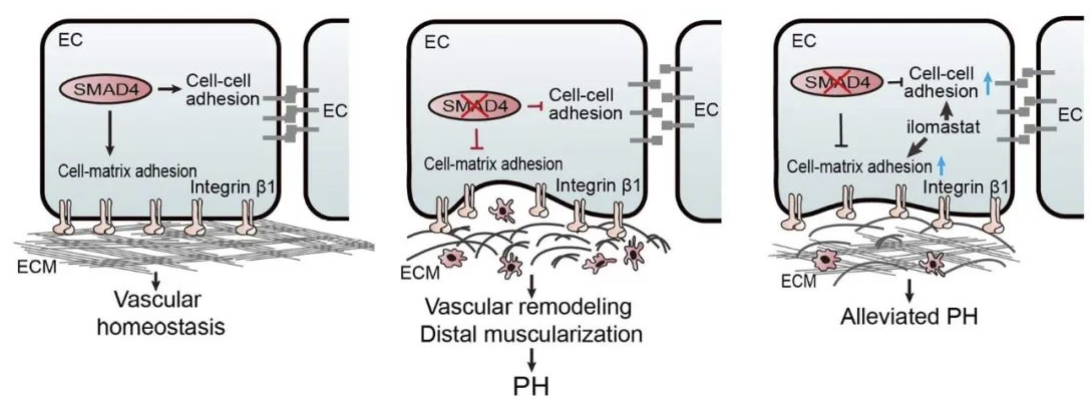
研究亮点三:机制验证与潜在治疗靶点探索
为进一步验证“细胞黏附障碍驱动肺高压”的假说,研究团队构建了内皮细胞中特异性敲除Integrin β1的小鼠模型,发现该模型再现了SMAD4缺失动物的肺高压表型。这一发现直接确认了Integrin介导的黏附功能在PH发病机制中的关键作用。
此外,研究者还发现,通过注射基质金属蛋白酶(MMP)抑制剂,可增强基底膜与ECM稳定性,从而有效延缓或逆转SMAD4缺失引起的PH进展。这一干预策略为开发靶向改善内皮黏附和基质完整性的治疗方法提供了重要实验基础。
结语:探索内皮稳态新通路,开启肺高压精准治疗新篇章
综上所述,该研究首次从分子与结构层面揭示了内皮SMAD4缺失如何通过影响细胞连接与基质稳态,驱动肺动脉高压的发生。不仅加深了对PH发病机制的理解,也为未来开发针对内皮功能重建的新型治疗手段提供了理论支持。
参考文献:
https://www.ahajournals.org/doi/10.1161/HYPERTENSIONAHA.124.22782

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言











#肺动脉高压#
12