
四代EGFR-TKI:应对非小细胞肺癌C797S突变等耐药机制的新希望
2025-01-03 苏州绘真医学 苏州绘真医学 发表于陕西省

第四代 EGFR TKI 的开发受到极大关注,因为这些药物有可能抑制第三代 EGFR TKI 耐药 NSCLC 患者中常见的耐药突变。
在全球范围内,非小细胞肺癌(NSCLC)占肺癌病例的 80% 以上,死亡率很高,然而,靶向常见表皮生长因子受体(EGFR)突变(即 del19、L858R)为 NSCLC 的治疗提供了范式转变。然而,罕见 EGFR 突变对 EGFR 靶向药物的敏感性存在差异,取决于具体的外显子 18-21 分子改变,其潜在的生物学机制尚不清楚。外显子 18 G719X、外显子 21 L861Q 、外显子 20 S768I 和外显子 20 插入突变是罕见突变中最常见的突变。第四代 EGFR 酪氨酸激酶抑制剂(TKI)的开发受到越来越多的关注,因为这些药物能够抑制 NSCLC 患者中常见的第三代 EGFR TKI 耐药突变(例如 C797S)。BDTX-1535 是一种口服生物可利用、能穿透血脑屏障、突变选择性、不可逆的 EGFR 抑制剂,在 NSCLC 和胶质母细胞瘤中具有显著的抗肿瘤活性(I/II 期试验正在进行中)。它是第四代 EGFR 抑制剂,在临床前模型中可以克服对奥希替尼的耐药性,并在携带 C797S 突变的 NSCLC 患者中显示出有希望的活性。在实验模型中,BDTX-1535 抑制所有常见 EGFR 突变和 50 多种罕见突变,包括 T790M、C797S、L718X、E709X、S784F、V834L 和 A289V,然而,外显子 20 插入受到的抑制程度要小得多。此外,EGF 受体细胞外结构域突变(例如 EGFRvII、III、IV)也可以被阻断。应该注意的是,在奥希替尼或其他 EGFR TKI 治疗后进展的所有 NSCLC 患者中,高达 50% 无法确定潜在的耐药机制,提示非突变信号转导通路也可能起作用,已发现肿瘤内异质性是耐药的重要因素,包括 3 种主要机制:(I)耐药持久性(DTP)细胞,(II)染色体不稳定性,和(III)染色体外 DNA(ecDNA)(见于超过 50% 的 NSCLCs),提示新型 EGFR TKI 在充分靶向在靶耐药机制方面将包括许多挑战。因此,在携带 C797S 突变及其他耐药机制的 NSCLC 患者中,迫切需要开发能够克服 TKI 耐药的新型药物。
背 景
在全球范围内,非小细胞肺癌(NSCLC)占肺癌病例的 80% 以上,并且死亡率很高。在中国,它也是最常见的癌症,也是癌症相关死亡的主要原因之一。靶向表皮生长因子受体(EGFR)改变为 NSCLC 的治疗提供了范式转变。在欧洲和美国,EGFR 突变占所有 NSCLC 病例的 10-20%,在亚洲占超过 45%。外显子 19 缺失和外显子 21 L858R 突变是最常见的改变,约占 NSCLC 突变的 90%;这些被称为“经典”(常见)突变,导致对酪氨酸激酶抑制剂(TKI)治疗高度敏感。
值得注意的是,在外显子 19 缺失和外显子 21 L858R 突变的 NSCLC 患者中,TKI 治疗的无进展生存期(PFS)长于铂类化疗。其他 EGFR 突变被称为“罕见”突变,占所有 EGFR 突变的 18%(图 1)。
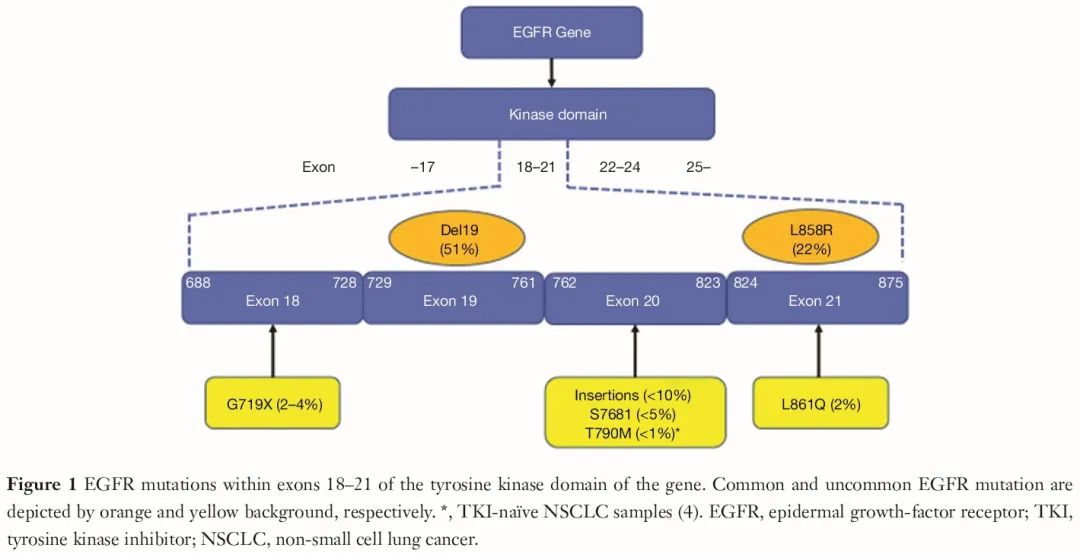
图1
罕见EGFR突变
研究发现,罕见 EGFR 突变对 EGFR 靶向药物的敏感性存在差异,取决于具体的外显子 18-21 分子改变,目前未得到充分研究。外显子 18 G719X、外显子 21 L861Q、外显子 20 S768I 和外显子 20 插入(例如,S768_D770dup、A767_V769dup、N771_H773dup、P772_H773insV、p.A767delinsASVD)突变通常被认为是罕见突变中最常见的改变。
已知外显子 20 插入突变对先前获批的 EGFR 靶向药物耐药,目前,铂类化疗和免疫检查点抑制剂治疗是罕见 EGFR 突变 NSCLC 患者的标准治疗。Wu 等人首次表明,携带外显子 20 插入的 NSCLC 患者的 PFS 显著短于外显子 19 缺失和外显子 21 L858R 突变的患者(1.4 个月vs 8.5 个月,P<0.001),提示靶向罕见突变的新药开发是一项高度未满足的医疗需求。
EGFR突变在其他癌症中的作用
EGFR作为药物新靶点在成人癌症中已得到大量研究,但在儿童中的研究较少。在多种儿童肿瘤中发现了 EGFR 通路改变(例如,EGFR 扩增或过表达),然而,复发性疾病的治疗结局仍然很差。此外,探索靶向 EGFR 的 TKI 治疗儿童患者的研究表明,几乎没有活性,这可能是由于绕过了与 EGFR 以外的其他因素相关的致癌信号转导(例如 c-MET)。
EGFR 在人类癌症中经常发生突变,并且在携带某些 EGFR 改变的 NSCLC 患者中是有效的治疗靶标。EGFR 突变主要发生在受体细胞内酪氨酸激酶结构域,从而直接促进其活性。
在过去的二十年中,一些研究表明,在儿童脑肿瘤(例如胶质母细胞瘤)中发现了某些 EGFR 突变或扩增,然而,这些突变都在细胞外,它们作为驱动突变的作用远不清楚。它们包括外显子 20小框内插入(胞内结构域),或错义突变(外显子 7 胞外配体结合结构域),可能不伴随基因扩增。在这些已报道的 EGFR 突变中,最常见的是 EGFRvIII。这种突变的特征是胞外结构域中 267 个氨基酸缺失,形成无法结合 EGF 配体且永久活性的突变受体。EGFRvIII 和其他细胞外变异被认为是晚期事件,主要发生在野生型 EGFR 扩增后。
第四代EGFR TKI
BDTX-1535(图 2)是一种口服生物可利用、能穿透血脑屏障、突变选择性、不可逆的 EGFR 抑制剂,在 NSCLC 和胶质母细胞瘤中具有显著的抗肿瘤活性。它是第四代 EGFR 抑制剂,在临床前模型中可以克服对奥希替尼的耐药性,并且在携带 C797S 突变的 NSCLC 患者中显示出有希望的活性。值得注意的是,BDTX-1535 很容易穿透血脑屏障,因此可能对治疗 NSCLC 中枢神经系统(CNS)转移有很大益处。此外,BDTX-1535 在胞外结构域突变的胶质母细胞瘤中具有高活性。
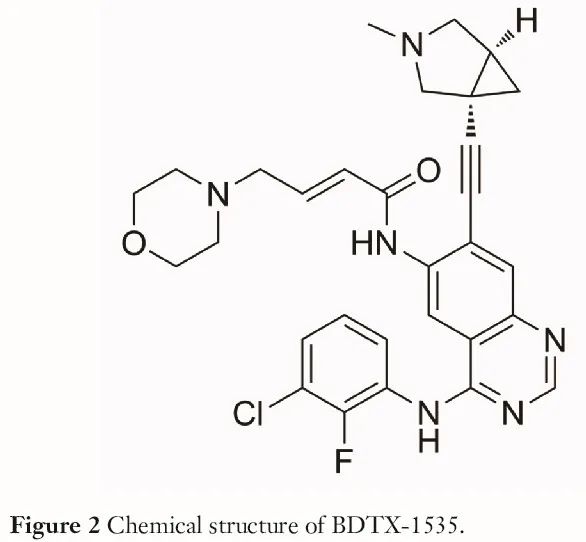
图2
实验模型显示,BDTX-1535 抑制所有常见 EGFR 突变和 50 多种罕见突变,包括 T790M、C797S、L718X、E709X、S784F、V834L 和 A289V,然而,外显子 20 插入受到的抑制程度要小得多。此外,EGFR 细胞外结构域突变(例如,EGFRvII、III、IV;图 3)也可以被阻断。
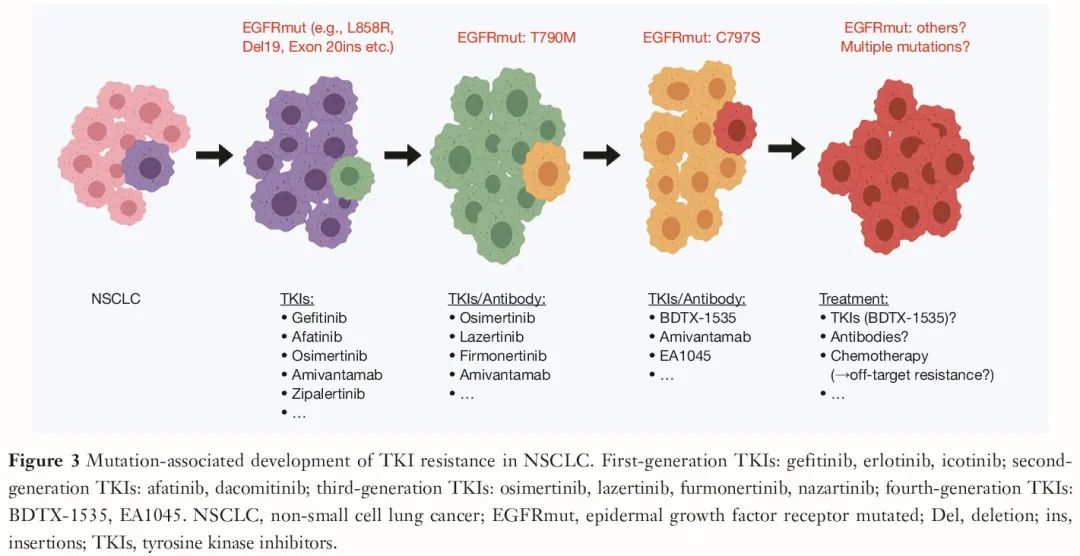
图3
对于携带常见 EGFR 突变的 NSCLC 患者,随着一线治疗中可逆的 EGFR TKI(第一代)被新型第三代 TKI(例如奥希替尼)替代,目前,在获得性耐药突变中,T790M 突变减少,C797S 突变出现且增加(占 EGFR 突变患者的 5-15%)。应该注意的是,许多携带 L858R 突变的 NSCLC 患者也携带罕见突变,这可能至少部分解释了为什么携带 L858R 突变的患者对奥希替尼的敏感性低于外显子 19 缺失患者。对于后者,同时存在罕见突变的情况较少,这些患者更常发生 C797S 突变作为获得性耐药机制。
一项 I/II 期研究正在探索 BDTX-1535,对 NSCLC 和胶质母细胞瘤分别进行剂量扩展和剂量递增试验(NCT05256290)。外显子 20 插入的患者不符合入组条件。27 例 NSCLC 患者的初步结果显示,BDTX-1535(100mg,每日一次,口服)在几乎所有 EGFR 突变的 NSCLC 患者中都具有活性,包括 T790M、C797S、L747P、L718Q 以及复合突变。即使在最高剂量水平(每日 200mg)下,该药物也只有轻度或中度不良事件。在这些既往接受过多线治疗的人群(既往治疗线数:1-9)中,客观缓解率(ORR)为 36%(22 例可评估患者),大多数患者获得持久缓解。此外,6 例患者在 ≥100mg,每日一次的剂量下,疾病稳定 [疾病控制率(DCR)85%]。19/22 例患者在入组前对奥希替尼耐药。在这 19 例患者中,ORR 为 42%,截至数据截止时,14 例患者仍在接受治疗。最常见的不良事件是皮疹(70%,2 例患者 3 级,无 4 级)和腹泻(35%,未见 ≥3 级)。
迄今为止,几种用于治疗 NSCLC 的抗 EGFR TKI 和单克隆抗体已获批 [欧洲医学协会(EMA)、美国食品和药物管理局(FDA)] 或处于后期临床开发阶段(表 1)。其中,仅有两种在临床试验中能以较低的半数抑制浓度(IC50)值靶向 C797S 突变:tigozertinib(以前称为 BLU-945)(IC50:2.9-4.4 nM)和 BDTX-1535(IC50:<10 nM)(表 2)。tigozertinib 的开发于 2024 年 1 月停止,其姊妹化合物 BLU-701 的临床开发也因无效而停止(表 2)。EAI-045 是首个变构激酶抑制剂;该分子不与 Cys797 结合,因为其残基位于变构结合口袋之外,但是,发现该药物具有显著更高的 IC50(0.33 μM),目前尚未进行临床评估。
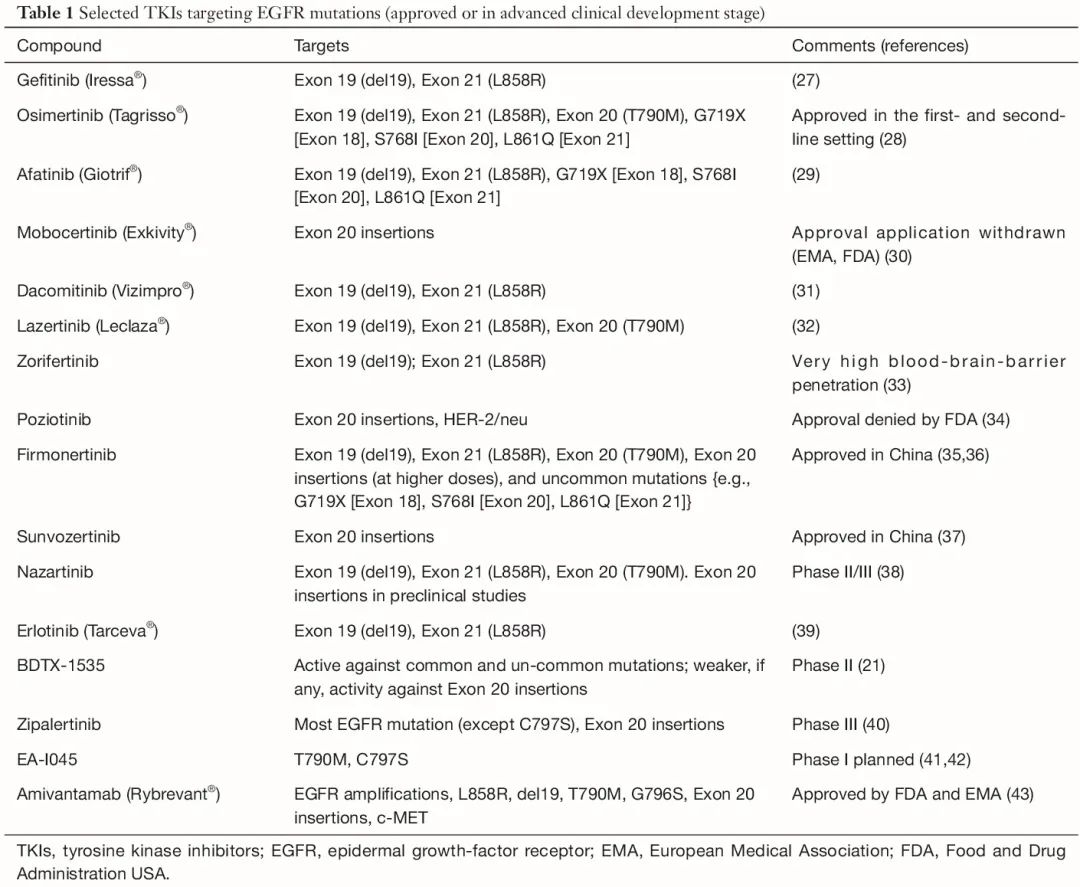
表1
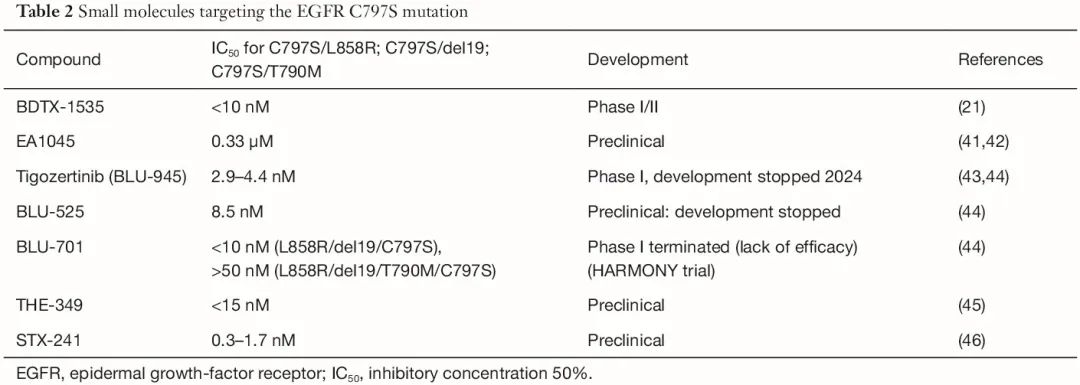
表2
相比之下,BDTX-1535 是一种泛 EGFR 突变 TKI,靶向常见和罕见 EGFR 突变(包括 C797S)以及细胞外变异和 EGFR 扩增。然而,特别有趣的是,BDTX-1535 是首个处于临床开发、也可以靶向细胞外 EGFR 突变/变异和扩增的分子,这可能对其他肿瘤实体(例如胶质母细胞瘤)有益。有趣的是,尽管 BDTX-1535 的激酶谱很广,但似乎对携带外显子 20 插入的 NSCLC 患者只有较弱的活性,因此,这些患者没有被纳入正在进行的临床试验。由于外显子 20 插入包含一组非常异质和多样化的 EGFR 改变,包括移码和非移码基因突变,因此可以想象这些患者中没有特异性 BDTX-1535 结合位点。
EGFR外显子20插入
目前只有少数化合物可用于治疗携带外显子 20 插入的 NSCLC 患者(表1)。其中,埃万妥单抗、伏美替尼、舒沃替尼和 zipalertinib 是临床开发中进展最为领先的化合物。埃万妥单抗是一种靶向 EGFR 和 c-MET 的双特异性单克隆抗体,被 EMA 批准用于铂类治疗后的二线治疗,被 FDA 批准与铂类和培美曲塞联合用于一线治疗,或既往铂类化疗后的二线治疗。结合后,抗体-受体复合体被蛋白酶体内化和降解(包括包含 T790M、C797S 和其他突变的胞内结构域)。
因此,在这种情况下,将靶向外显子 20 插入的特异性 TKI 与 BDTX-1535 相结合的未来临床试验可能对某些 NSCLC 患者有益。
C797S突变再敏感方法
EGFR 基因 C797S 突变介导对目前所有可及的第三代 TKI 的耐药性。已知 C→S 突变(即 C481S)也会导致对伊布替尼的耐药性,提示 C→S 突变可能是一种重现性关键突变,可阻碍抑制剂与多种酪氨酸激酶的结合。使用这些新化合物治疗较长时间后,很可能会再次出现耐药克隆。随后是否会检测到新的 EGFR 突变,或者是否出现一组不同的突变共同导致耐药,至少在一定程度上与间变性淋巴瘤激酶(ALK)抑制剂的耐药发生情况类似,还有待观察。最后,也可以想象 C797S 抑制剂可能会越来越多地引发脱靶耐药机制,提示这些患者届时可能只能接受化疗(图 3)。
最近发表的研究提供了越来越多的证据表明,EGFR T790M 和 C797S 突变的等位基因关系似乎对 EGFR 靶向 TKI 的反应至关重要。在这方面,已经表明,携带T790M/C797S反式突变(在不同 DNA 链上)的 NSCLC 患者对第一代和第三代 EGFR TKI(例如吉非替尼和奥希替尼)联合疗法仍然敏感。然而,如果这些突变处于顺式等位基因关系(在同一 DNA 链上),则没有 TKI 单药或联合疗法可以抑制耐药克隆,提示 C797S 从反式到顺式的克隆进展可能是新的耐药机制。因此,可以想象,克隆异质性可能起重要作用,并且可能同时存在于奥希替尼耐药肿瘤中。另一方面,反式等位基因关系的识别可能为基于 C797S 的奥希替尼耐药的 NSCLC 患者提供个体化治疗机会。
C797S以外的耐药机制
应该注意的是,在奥希替尼治疗后进展的所有 NSCLC 患者中,高达 50% 无法确定潜在的耐药机制,提示非突变信号转导通路也可能起作用。在这方面,Haratake 等人最近首次证明,致癌蛋白 MUC1-C(MUCIN)是 NSCLC 患者获得性奥希替尼耐药的重要驱动因素,支持奥希替尼耐药机制多样,因此开发能够克服这种临床上观察到的耐药的治疗策略具有挑战性。已知 MUC1-C 与多种癌基因一起作用,驱动癌症干细胞、DNA 损伤抵抗和免疫侵袭。Haratake 等人使用奥希替尼耐药细胞系 H1975-OR 和 MGH700-2D 细胞(来源于奥希替尼耐药 NSCLC 患者),证明奥希替尼耐药与 MUC1-C 水平升高有关,并且使用GO-203抑制MUC1-C可在人源异种移植模型中恢复敏感性。Hu等人提供了类似的结果,他们证明在患者来源的小鼠异种移植模型中,奥希替尼耐药性与神经内分泌谱系转录因子 achaete-scute 同源物 1(ASCL1)上调有关。ASCL1对于大多数具有神经内分泌特征的肺癌的生存至关重要,ASCL1启动上皮 - 间充质基因表达程序,导致奥希替尼耐药的发生。
肿瘤异质性和EGFR TKI耐药
除了靶向 C797S 突变外,肿瘤异质性和几种耐药机制的共同表达可能是克服临床观察到的奥希替尼耐药性的巨大障碍。
此外,肿瘤内异质性被认为对 NSCLC 患者 EGFR TKI 耐药有重要影响。在单个肿瘤中存在许多肿瘤亚克隆。对于 NSCLC 患者,已知耐药持久性(DTP)细胞克隆能够通过中性选择加速药物治疗耐药性的发生。
瘤内异质性定义为观察到可能保留少数 TKI 耐药 NSCLC 细胞群克隆,然后,这些克隆重新开始增殖,导致 NSCLC 复发或进展,即使大多数 TKI 敏感细胞对治疗有反应。它包括三个主要机制:(I)DTP 细胞,(II)染色体不稳定性,以及(III)染色体外 DNA(ecDNA)。普遍认为,ecDNA 是肿瘤遗传异质性的主要促成因素,见于 50% 的 NSCLC。EGFR 突变以外的基因改变也可能导致 DTP 状态。
NSCLC 细胞会经历各种变化来适应 TKI 治疗引起的新肿瘤微环境(TME)。DTP 细胞被认为在此过程中起关键作用,并且可能是观察到的耐药机制的关键参与者。此外,据推测,在 DTP 状态期间会获得特定的获得性染色体改变(例如,T790M、C797S、c-MET 扩增等)。观察到 DTP 细胞的存活由 H3K9 甲基化和 H3K27 甲基化导致,强调了 DTP 对肿瘤耐药的重要性。此外,参与 DNA 损伤修复的多梳抑制复合体 2(PRC2)可通过 zeste 同源物 2 增强子(EZH2;PRC2 的酶亚基)使H3K27发生甲基化,提示抑制 EZH2 活性可以抑制肺癌 DTP 细胞克隆。
总 结
第四代 EGFR TKI 的开发受到极大关注,因为这些药物有可能抑制第三代 EGFR TKI 耐药 NSCLC 患者中常见的耐药突变(例如 C797S)。然而,尽管临床活性有希望,但这些化合物的开发在充分靶向在靶耐药机制方面仍面临一些障碍。主要障碍之一是 TKI 耐药的发生可能由脱靶突变或其他信号通路旁路激活引起。此外,将第四代 EGFR TKI 与其他治疗方式(例如免疫疗法或其他 EGFR TKI)联用的理想策略尚未确定,需要进行更多的临床前和临床研究,以更好地了解其管理肿瘤内异质性的能力。解决 C797S 突变以外的治疗耐药性将是一个主要目标,结合我们对 DTP 细胞和 ecDNA 对肿瘤内异质性影响的知识可以为 NSCLC 患者带来更有效的策略。全面基因组测序的使用现在已成为临床标准,通过精准医学和人工智能的进步,可以加速对耐药机制的更深入理解。更好地了解 NSCLC 耐药的分子因素可能为开发用于治疗 NSCLC 的新型、个性化、分子靶向药物铺平道路。迫切需要开发能够克服携带 C797S 突变及其他耐药机制的 NSCLC 患者 TKI 耐药的新型药物。如果这些结果可以在更大规模的随机试验中得到证实,将对未来 NSCLC 患者的治疗产生重大的临床意义。
参考文献:
Dempke WCM, Fenchel K. Targeting C797S mutations and beyond in non-small cell lung cancer-a mini-review. Transl Cancer Res. 2024 Nov 30;13(11):6540-6549. doi: 10.21037/tcr-24-690. Epub 2024 Nov 27. PMID: 39697713; PMCID: PMC11651777.

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言










#非小细胞肺癌# #EGFR-TKI#
12