
【Blood】综述:成人B-ALL的基因亚型
2025-01-16 聊聊血液 聊聊血液 发表于陕西省

《Blood》近日发表综述,概述了成人B-ALL的基因亚型,包括已明确亚型中的最新生物学和临床见解以及新亚型的数据,还讨论了它们与风险分类、疾病监测和治疗的相关性。
成人ALL基因分型
B细胞急性淋巴细胞白血病(B-ALL)是一种罕见的成人恶性肿瘤,其预后仍然很差,特别是与儿童相比。在过去的二十年中,广泛的全基因组研究已经确定了许多驱动白血病的遗传改变,从而识别出20多种不同的亚型,这些亚型与治疗反应和预后密切相关。在儿童B-ALL中,大量相关研究使得遗传分类成为风险适应性(risk-adapted,或称根据风险调整治疗)治疗策略的核心组成部分。但基因亚型在不同年龄中分布并不均匀,且成人B-ALL的基因改变特征及其预后相关性的研究较少,治疗主要基于是否存在BCR::ABL1融合。
《Blood》近日发表综述,概述了成人B-ALL的基因亚型,包括已明确亚型中的最新生物学和临床见解以及新亚型的数据,还讨论了它们与风险分类、疾病监测和治疗的相关性。
引言
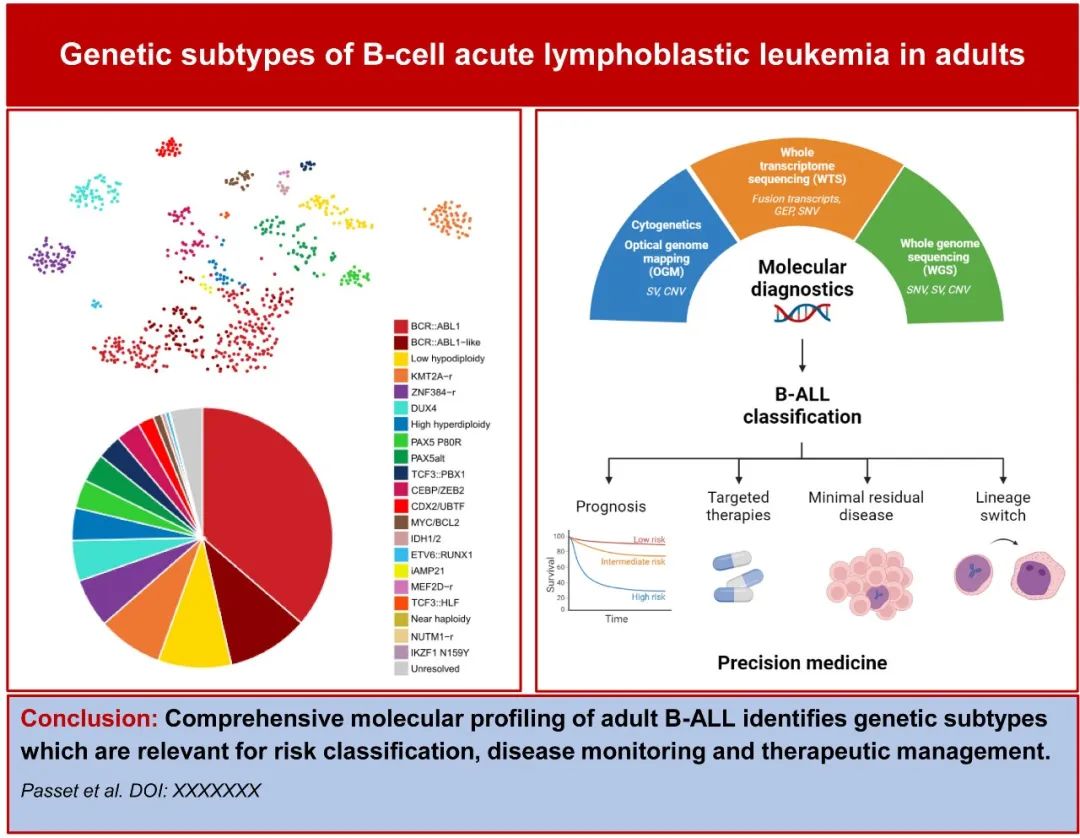
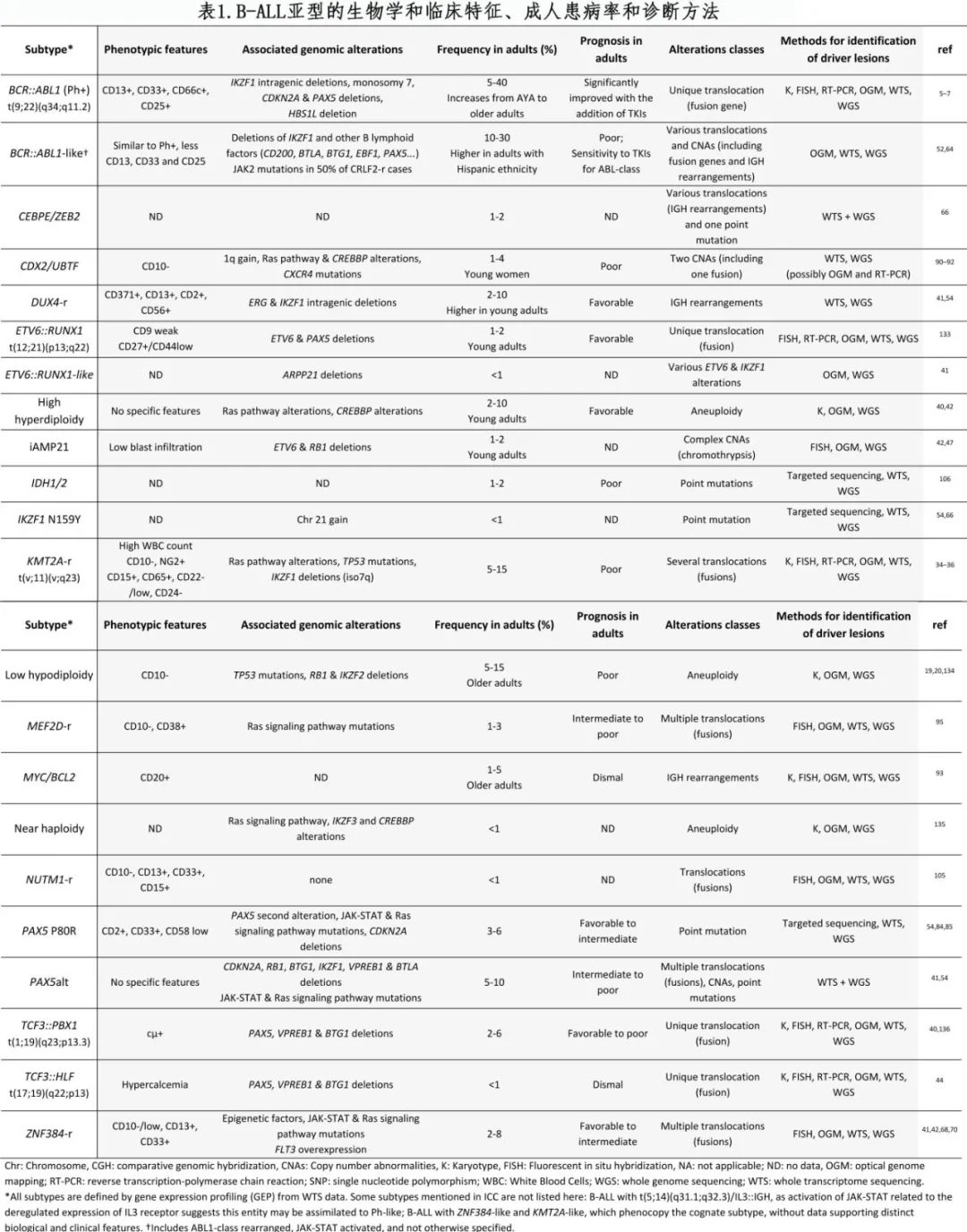
B 细胞急性淋巴细胞白血病 (B-ALL) 是由多种基因改变驱动的,这些基因改变定义具有不同致癌通路和生物学特征的不同亚型,且这种异质性在预测预后和分层治疗方面具有很强的临床意义。长期的细胞遗传学特征发现了复发性染色体易位和特殊的非整倍体,但大部分问题仍未结局。从21世纪初开始,使用微阵列和全转录组测序 (WTS) 对大型 B-ALL 队列进行的基因表达分析,通过与不同的基因表达谱匹配加强了所谓的“驱动性”或“主要”基因改变所定义的基因亚型的意义;这些方法也发现了由各种复杂分子异常定义的新型 B-ALL 亚型。因此,国际共识分类 (ICC) 承认26种不同的分子亚型和其他临时实体(表1),需要在常规诊断检查中实施深入的基因表征。从儿童到青少年、年轻和老年人,B-ALL基因亚型的流行并不均匀,从而解释了该疾病在不同年龄人群之间预后的差异。
历史上的细胞遗传学亚型的新见解
费城染色体(Ph)-阳性 (BCR::ABL1)
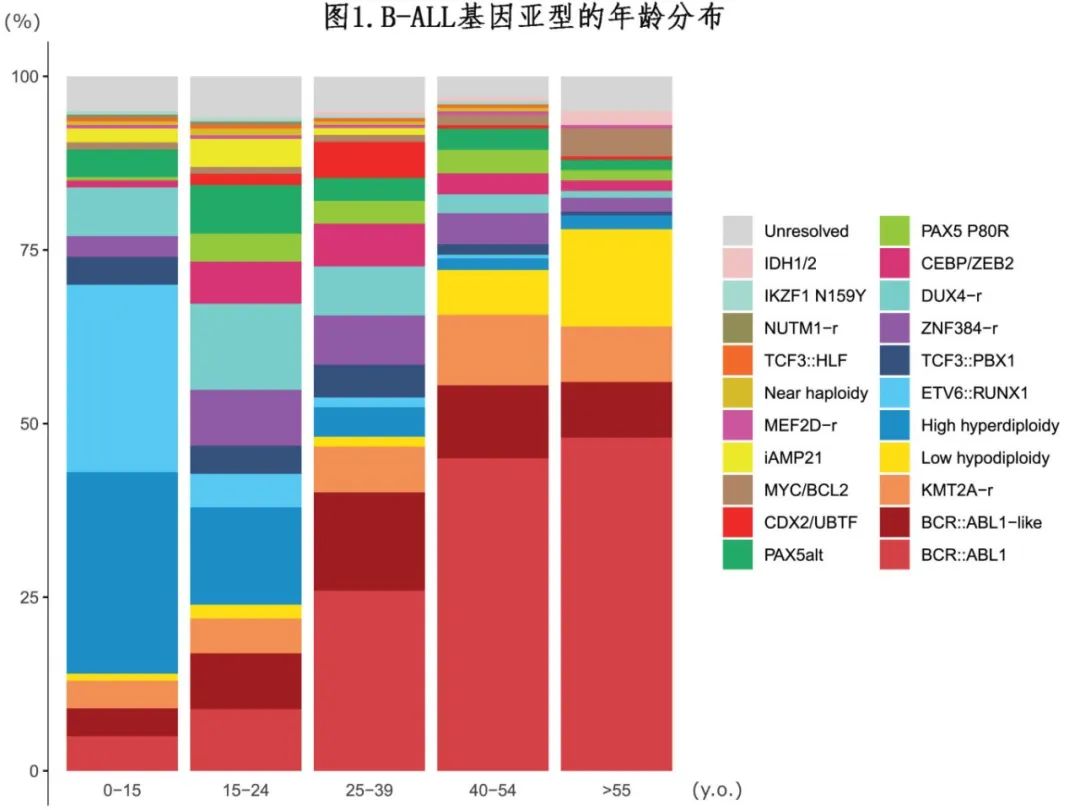
t(9;22)(q34;q11) 易位产生BCR::ABL1融合基因,从而定义了成人B-ALL 中最常见的基因亚型(图1)。其发病率随年龄增长而增加,>55岁 B-ALL中高达一半。费城染色体阳性 (Ph +) ALL对化疗反应差,历史上预后不佳,而加入酪氨酸激酶抑制剂 (TKI) 的方案和贝林妥欧单抗可显著改善预后长期生存率已接近80%。鉴于此,正在重新评价化疗和异基因造血干细胞移植 (allo-HSCT) 的作用。
最近的研究揭示了Ph+ ALL内显著的生物学异质性,具有很强的临床意义。首先,特定共突变可影响预后,IKZF1plus模式(即 IKZF1 缺失伴 PAX5 或 CDKN2A 缺失)为高危。其次,BCR::ABL1在部分患者的非淋巴母细胞中得到证实,从而定义了介于单纯淋巴母细胞Ph+ ALL和慢性髓性白血病 (CML)的实体(图2)。
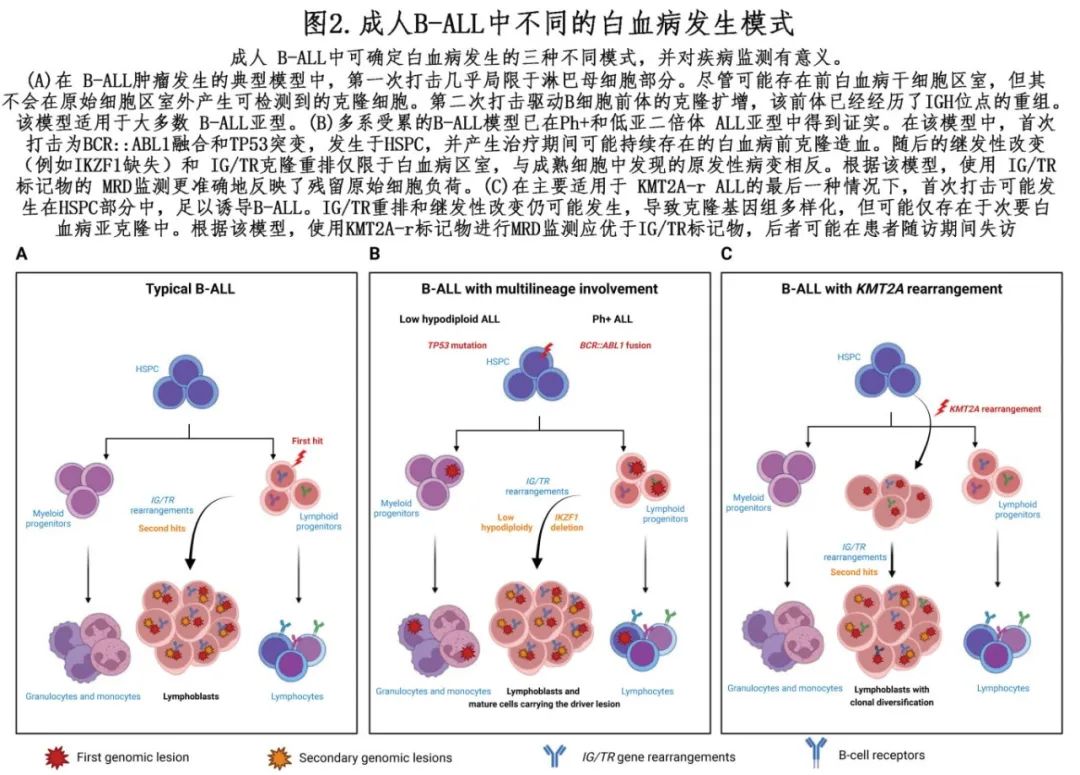
这种“CML样”或“多系”Ph+ ALL占de novo Ph+ ALL患者的32-37%。因此使用免疫球蛋白或 T 细胞受体 (IG/TR) 基因重排(而非BCR::ABL1)定量监测可测量残留病 (MRD),可更好地反映残留淋巴母细胞负荷,并更准确地预测结局。因此共突变和基于 IG/TR 的 MRD 正在成为风险分层和治疗决策的标志物。
最后,转录组学研究显示了与 B 细胞分化阶段、谱系受累和共突变相关的不同Ph+ ALL特征。简言之,一个cluster显示早期 pro-B 特征,这是一种独特的共突变模式,包括 HBS1L 缺失、单体7和参与 B 细胞定型(commitment)和分化早期阶段的转录因子(即RUNX1、MEF2C、EBF1)改变,并与多系受累相关。另一个cluster类似于晚期前 B/前 B 细胞,富含 IKZF1 和 CDKN2A 双等位基因缺失和 PAX5 缺失。在该组中,BCR::ABL1仅限于淋巴部分。虽然该分类的预后意义还需要进一步验证,但发育特征可能提供额外的见解,例如在 B 细胞靶向药物后白血病倾向于转换为髓系细胞,或存在能够产生新型Ph+ ALL的前白血病储存库。
低亚二倍体
低亚二倍体(Low hypodiploidy) ALL 在细胞遗传学上定义为30-39条染色体的核型,长期以来被认为是一种与不良预后相关的罕见亚型。在某些情况下,低亚二倍体细胞的全基因组可复制产生超二倍体基因组(称为掩盖或重复的低亚二倍体),存在诊断挑战,并可能导致低估其真实发生率。最近的基因组研究显示,成人中的患病率相对较高,并随着年龄的增长而增加,在老年Ph 阴性 B-ALL 中患病率高达30%。值得注意的是,近单倍体(near-haploidy;24-29条染色体)是一种独特的实体,在成人中非常罕见。TP53双等位基因异常通常包括一个突变和一个与17号单体相关的拷贝丢失,是低亚二倍体的标志。在半数儿童病例中发现胚系 TP53 突变,表明其早于导致白血病的非整倍体(aneuploidy)事件。成人呈体细胞多系受累,表明 TP53 突变的克隆性造血 (CH) 可能是低亚二倍体 ALL 的易感条件。因此,TP53突变 CH 的年龄相关增加可以解释低亚二倍体 ALL 的患病率随年龄增长而增加,以及其作为治疗相关白血病的发生。重要的是,由于 TP53突变 CH 也可进展为髓系恶性肿瘤,可能影响缓解后治疗策略和患者监测。
KMT2A重排
在各种急性白血病中均可观察到 KMT2A 重排,或称t(v;11)(v;q23),其伴侣基因具有相对谱系特异性。在 B-ALL 中,t(4;11)(q21;q23)导致的KMT2A::AFF1融合占 KMT2A 重排 (KMT2A-r) 病例的80%以上。虽然 KMT2A-r 在婴儿白血病中普遍存在,但其在儿童中罕见,但在成年期变得更常见,并且可能与既往恶性肿瘤的细胞毒治疗相关。KMT2A-r在儿童和成人中均与不良预后相关,但相关生物学和治疗开发的研究集中于婴儿。在成人中,KMT2A-r ALL患者适合allo-HSCT,尽管预后仍不理想。近期研究强调了特定共突变的预后重要性,即 TP53 点突变和 IKZF1 缺失,以及诱导后MRD,能够识别具有不同预后的不同 KMT2A-r ALL 亚组。值得注意的是,缺乏不良因素的患者在单独强化疗后表现出极佳预后,挑战了 allo-HSCT 在该亚型中的系统使用;具有高危标准的患者预后不佳,需要替代疗法。鉴于 B 细胞靶向治疗有可能被这些白血病细胞的内在可塑性所规避(见“B细胞靶向治疗时代的预后意义”一节),应积极开发新的方法,如 menin 抑制剂。
其他细胞遗传学亚型
高超二倍体(High hyperdiploidy)以非随机染色体获得为特征,是最常见的儿童 B-ALL 亚型,但成人中不足10%。有趣的是,这种非整倍体源于初始有丝分裂错误而非染色体不稳定性。导致ETV6::RUNX1融合的隐匿性t(12;21)(p13;q22) 易位是第二常见的儿童实体,但其在成人中非常罕见 (< 2%),并且通常仅限于青少年和年轻成人 (AYA)。两种亚型在儿童中均高度可治愈,长期生存率超过95%;成人中尽管数据有限,但也可能与更好的预后相关。因此,成人和儿童之间高危亚型(例如Ph+、低亚二倍体和KMT2A-r)和低危亚型(例如高超二倍体,ETV6::RUNX1)的患病率差异在很大程度上解释了相反的临床结局(图1,表1)。
t(1;19)(q23;q13) 易位可导致TCF3::PBX1融合,约占儿童和成人 B-ALL 病例的4%,常与中枢神经系统受累相关。成人t(1;19)(q23;q13)的结局因治疗方案而异,介于差到好,最佳治疗方案(包括是否allo-HSCT)仍有争议。
t(17;19)(q22;p13) 易位可导致TCF3::HLF融合,定义了在儿童和 AYA 中发现的一种非常罕见的实体,预后不佳。但B细胞靶向治疗最近获得令人鼓舞的结果,临床前研究表明对维奈克拉和 BET 抑制剂敏感,但需要进一步的临床验证。
21号染色体的染色体内扩增(iAMP21) 是另一种罕见的高危实体,多见于儿童和 AYA,可获益于化疗强化。
转录组和/或全基因组测序揭示的新兴基因亚型
Ph样(BCR::ABL1样)
Ph 样 ALL 最初定义为类似Ph+ ALL但无BCR:::ABL1融合的基因表达谱,常见 IKZF1 缺失且对化疗反应差,并转化为预后不良。随后的 WTS 研究发现涉及细胞因子受体或酪氨酸激酶的多种基因改变,导致细胞信号通路的异常激活,可能适合药物抑制。Ph样 ALL 发生在所有年龄组中,但在 AYA 中略微更常见,占B-ALL的30%。基因改变的极端多样性使得诊断具有挑战性,而WTS是识别基因表达谱和捕获全部原发病变的最有效方法。涉及 ABL 类基因(例如ABL1、ABL2、PDGFRA、PDGFRB、CSF1R)的融合占 Ph 样 ALL 的10-15%,当仅采用常规化疗治疗时,与频繁的诱导失败和不良预后相关。添加 TKI 显示临床获益,需要在诊断中进行 ABL 类融合筛查。还需要更多研究来完善基于受累激酶的 TKI 选择,并重新评估 allo-HSCT 的作用。在50%的 Ph 样 ALL 中发现 CRLF2 基因重排,包括IGH::CRLF2和P2RY8::CRLF2融合。IGH::CRLF2融合在成人中更常见,并且始终与 Ph 样特征相关,而P2RY8::CRLF2可作为非 Ph 样 ALL 的继发事件。常见的共突变包括 JAK2 激活突变和增强 JAK-STAT 信号转导的其他改变(例如 IL7RA 和 JAK1 激活突变、SH2B3功能丧失改变),以及IKZF1、PAX5和 CDKN2A 缺失。与 CRLF2 相似,EPOR重排(包括免疫球蛋白基因座的易位和隐性插入)可导致 EPOR 过表达和 JAK-STAT 通路激活。JAK 类 Ph 样亚型也与不良预后相关,强调了靶向治疗的价值。然而JAK1/2抑制剂芦可替尼单药治疗的活性有限,评价其联合化疗的 I/II 期研究结果仍有待确定 (NCT02723994、NCT02420717、NCT03571321)。
ZNF384-r
该亚型由与各种伴侣基因(包括EP300、TCF3、TAF15、CREBBP、ARID1B和 EWSR1)融合的 ZNF384 转录因子基因 (ZNF384-r) 的基因组重排所定义。所有这些融合保留整个 ZNF384 编码区,产生了共同的基因表达特征。其他 ZNF384 改变(例如突变和缺失)和涉及其类似物 ZNF362 的融合也有报道,表型为ZNF384-r(称为 ZNF384 样)。ZNF384-r ALL多表现出特征性免疫表型,包括 CD10 弱表达和髓系标志物 CD13 和/或 CD33 的异常表达,甚至是 B/髓系混合表型。来自小型成人队列的数据表明 ZNF384-r ALL 具有良好或中等预后。值得注意的是,该实体具有多向分化潜能和谱系可塑性,可在患者随访期间通过免疫表型转换表现出来。有趣的是,ZNF384融合蛋白驱动与 FLT3 抑制剂敏感性相关的 FLT3 基因表观遗传活化,或是该亚型的可行治疗选择。
DUX4-r
该实体最初通过与 ERG 基因内缺失的独特关联被识别,随后的 WTS 显示IGH::DUX4融合为初始改变,驱动 DUX4 的异位表达。转录因子 DUX4 的致白血病作用涉及与 ERG 基因座结合,导致具有显性负活性的替代 ERG 转录本的表达和由于 RAG 机制的可及性增加导致的频繁 ERG 基因内缺失。尽管 IKZF1 基因内缺失在该实体中很常见,但在几项儿童研究和有限的成人研究中与良好预后相关。识别 DUX4-r 病例具有挑战性,因为IGH::DUX4融合在细胞遗传学上为隐秘性,并且 WTS 检测不一致,而 ERG 缺失仅存在于三分之二的病例中。因此需要全面的基因表达谱或 WGS 来识别所有病例。有趣的是,DUX4-r ALL在诊断时或治疗开始后早期有时会出现单核细胞样细胞,显示来源于发生单核细胞转换的 B-ALL 克隆。单核细胞和 T 细胞标志物CD371、CD2和 CD56 在 B-ALL 细胞上的频繁异常表达也提示谱系混杂性。支持这一点的是,单细胞基因组学证实 DUX4-r B-ALL 原始细胞向髓系和 T 细胞系的多系启动。虽然这可能意味着逃避 B 细胞导向治疗的风险,但迄今为止尚未报告此类病例。
PAX5 P80R和PAX5alt
PAX5 是 B 细胞淋巴样分化的主要调节因子,是 B-ALL 中公认的抑癌因子。PAX5的基因组改变(包括缺失和功能丧失性突变)常继发于起始病灶(如BCR::ABL1融合)。然而PAX5p.P80R点突变为初始致癌事件,因为它不包括其他遗传驱动因素,与独特的基因表达谱相关,并且可以通过诱导非典型转录程序启动 B-ALL 的发生。该突变通常与染色体 9p 缺失导致的第二个 PAX5 等位基因的突变或丢失相关,也可导致 CDKN2A/B 位点的丢失。RAS(NRAS、KRAS、PTPN11和NF1)或JAK/STAT(IL7R、JAK2、PTPRC和SH2B3)信号基因突变很常见,从而突出了PAX5p.P80R在驱动白血病中的强烈致癌系统作用。PAX5 P80R B-ALL与成人中危至低危预后相关。
另一个称为 PAX5alt 的亚组在最新 ICC 中列为临时实体。它是通过基因表达聚类所确定,在基因组水平包括各种 PAX5 改变,包括基因融合、基因内扩增和p.P80R以外的突变。PAX5与ETV6、AUTS2、ELN和SOX5等伴侣融合基因先前被认为是启动白血病的驱动因素。值得注意的是,PAX5::JAK2融合可激活 JAK-STAT 通路,并具有 Ph 样特征。与 PAX5 融合不同,PAX5缺失和突变也可作为其他亚型的继发事件出现。因此,识别所有 PAX5alt 病例需要全面的基因组表征,包括检测点突变、拷贝数异常 (CNA) 和基因融合,以及基因表达特征。仅有很少的研究分析了 PAX5alt 组的预后,似乎介于中等到不良,但需要更多的数据来充分确立其临床意义。
CDX2/UBTF
该实体是通过大型成人 B-ALL 队列的基因表达分析发现的,这些基因表达分析发现一个小的未解析的cluster,随后的基因组研究发现了该亚型中唯一且普遍存在的两种新改变。第一种改变是13q12.2位点 RAG 介导的微缺失,靠近 CDX2 基因,包含 FLT3 和 PAN3 基因启动子。由于劫持 PAN3 增强子,该缺失显示诱导异位 CDX2 过表达。CDX2 是一种同源盒基因,通常在胚胎发育过程中表达,包括原始造血,并仅限于成人的肠道。第二种改变是17q21.3的微缺失,可导致 UBTF 和 ATXN7L3 基因融合,产生嵌合UBTF::ATXN7L3蛋白。UBTF参与核糖体 DNA 转录,而 ATXN7L3 是 SAGA 复合物的一个亚基,调节基因表达。这些改变所激活的致癌通路及其潜在的协同相互作用仍有待阐明。该 B-ALL 亚型主要影响年轻女性,在儿童中几乎未报告病例。CDX2/UBTF ALL存在独特的额外改变模式,包括 1q 重复和 CXCR4 突变。最重要的是,该亚型对治疗的反应非常差,导致高复发率,需要新型治疗方法。
MYC/BCL2
将癌基因 MYC 和 BCL2 分别置于 IGH 基因座转录控制下的重排(分别为t[8;14]和t[14;18])在成熟 B 细胞淋巴瘤中很常见。同时具有 MYC 和BCL2(和/或BCL6)重排的淋巴瘤称为“双打击”淋巴瘤,归为高级别 B 细胞淋巴瘤,与侵袭性疾病和极差预后相关。尽管在 B-ALL 中很少观察到这些改变,但当存在时,与同样较差的治疗反应和令人沮丧的预后相关。最近的一项研究强调了该组内的显著异质性,揭示存在 Burkitt 样白血病和真实 B-ALL 病例的混合。重要的是要评估哪些治疗策略(可能包括 B 细胞靶向治疗和 BCL2 抑制剂,它们在双打击淋巴瘤中有效)在这些病例中最有效。
MEF2D-r
涉及MEF2D(一种参与早期 B 细胞发育的转录因子)的基因组融合定义了另一种罕见的 B-ALL 亚型,最常见的融合伴侣为BCL9,但也报告了多个其他伴侣基因,如CSF1R、DAZAP1、FOX2、HNRNPH1和SS18。融合均保留MEF2D 的 MADS-box 结构域(对 DNA 结合至关重要),并导致 MEF2D 过表达。MEF2D-r B-ALL的特征为具有与 MEF2D 靶基因表达相关的独特基因表达谱。该B-ALL 亚型与中度至不良预后相关。值得注意的是,MEF2D过表达可导致 HDAC9活化,或可作为治疗靶点。临床前研究表明,组蛋白去乙酰化酶抑制剂可为治疗该亚型提供治疗获益。
CEBP/ZEB2和CEBPalt
IGH 基因座与 CCAAT 增强子结合蛋白 (C/EBP) 转录因子家族不同基因 (CEBPA、CEBPB、CEBPD、CEBPE、CEBPG) 之间的重排先前报告为 B-ALL复发性改变,不包括其他致癌驱动因子。C/EBP因子对造血系统的增殖和分化至关重要,其中 CEBPA 在正常髓系分化和急性髓系白血病中尤为重要;但C/EBP因子在淋系分化中的作用仍不清楚。涉及 CEB/P 基因的基因组重排很可能导致其在 B 细胞前体中的异常表达,从而驱动 B-ALL致癌程序。一种独特的转录谱与IGH::CEBPE融合和/或ZEB2 p.H1038R突变的存在相关,该突变在最新 ICC 中归类为临时实体。但也观察到 ZEB2 突变(包括p.H1038R)与其他亚型定义病变和低突变等位基因频率相关,表明这些突变可能作为协同作用而非通过特异性驱动子启动病变。需要进一步研究来阐明 C/EBP 和 ZEB2 改变的致癌作用及其临床相关性。最近一项关于唐氏综合征 (DS) 相关 ALL 的研究发现了另一种亚型,其特征为涉及不同 C/EBP 家族基因的重排、常见的基因表达特征和常见的 FLT3 激活突变。该亚型(称为C/EBPalt)未在非 DS 儿童队列中报告,也未纳入ICC,但需要进一步研究来确定其在成人 B-ALL 中的意义。
其他罕见和/或临时实体
最近发现了其他非常罕见的 B-ALL 实体,但关于其在成人中的患病率和临床意义的数据有限;罕见B-ALL包括在婴儿和儿童中观察到的由 NUTM1(NUTM1-r) 重排定义的亚型,以及另一种由IKZF1 p.N159Y突变定义的亚型;ICC还包括作为临时实体ETV6::RUNX1样、KMT2A-r样和 ZNF384-r 样,定义为与相关实体相似但缺乏典型重排的基因表达谱。报告的病例非常少,几乎没有成人数据。
近期研究发现了 B-ALL 中IDH1 p.R132C和IDH2 p.R140Q的热点突变,与髓系恶性肿瘤中已知的突变相同。这些突变与独特的基因表达特征和 B-ALL 原发改变的缺失有关,表明 IDH1/2 突变可能作为起始事件并定义成人中独特的 B-ALL 亚型,尽管未纳入ICC。重要的是,考虑到携带 IDH1/2 突变的 B-ALL 与不良预后相关,可能需要在这种情况下评价 IDH1/2 抑制剂的潜在获益。
继发性异常的作用
虽然亚型定义病变主要驱动 B-ALL 中的异常转录程序,但继发性改变也在白血病发生中发挥关键作用。B-ALL的继发性改变包括复发性 CNA和影响 B 淋巴因子(例如IKZF1、PAX5、EBF1)、细胞周期调节因子(例如CDKN2A、TP53)、细胞信号通路(例如NRAS、KRAS、FLT3、JAK2)、表观遗传因素(例如CREBBP、SETD2、KMT2D)的点突变。一些改变与耐药直接相关,并且可以在复发时获得(例如NT5C2、NR3C1);重要的是它们与原发性病变的相关性并不均匀,可能是由于原发性病变施加的强烈演变所限制。继发性改变可影响 B-ALL 的表型特征、治疗反应和临床预后,但建立稳健的预后相关性具有挑战性,其中一个原因在于继发性改变的频率和预后影响可能因潜在的基因亚型而存在显著差异。因此,除不同的治疗方案外,不同的队列(例如年龄、B-ALL亚型分布、基因起源)也可能得出不同的结论;例如许多研究探索了 IKZF1 改变对预后的影响,但结果不一致,尤其是在成人 B-ALL 中。由于 IKZF1 缺失在Ph+ 和 Ph 样ALL(较少)中较为常见,因此预后意义可能受到包括或不包括Ph+ ALL在内的队列组成和方法学(在多因素或亚组分析中考虑或不考虑原发病变)的显著影响。分析不同亚型内继发性改变的影响可能提供有价值的见解,此外还可以检查继发性改变的特定组合,因为它们可能具有预后意义,正如IKZF1+模式的影响所示。
临床实践中诊断的挑战
虽然大多数新的基因组见解都是通过对大型队列的库存标本进行高通量测序实现的,但实施精准医疗需要诊断实验室有可行的分子学方法,且存在与通量、周转时间和成本相关的限制。历史上 B-ALL 亚型的特征为染色体易位或非整倍体,使得标准细胞遗传学成为诊断检查的核心,随后辅以荧光原位杂交 (FISH) 和靶向分子检测 (RT-PCR、MLPA、LDA) 等技术检测复发性基因重排。然而已证明 B-ALL 的基因组格局是如此复杂,涉及许多基因和改变类型,以至于这些方法变得劳动密集型和低效。最近开发的光学基因组图谱 (OGM) 似乎是一种相对简单的实施技术,可通过检测大多数基因组重排(CNA和SV)(而非突变)克服常规细胞遗传学的部分局限性。因此全基因组测序 (WGS) 是对基因组进行详尽表征的唯一方法,但其仍较为昂贵且资源密集,使大多数诊断实验室目前无法使用该技术。最后,WTS具有将基因表达分析与重排和突变的检测相结合的独特优势,已经开发了预测工具以促进亚型分配,而不需要内部数据;其比 WGS 更容易访问,此外高通用性可能使其成为新的护理标准。
MRD监测的考虑因素
除基因改变外,MRD是 B-ALL 预后和治疗分层的关键基石。MRD分子标志物(包括 IG/TR 基因的克隆性重排和特异性基因变异)与各 B-ALL 亚型的致白血病过程和发育阶段密切相关。例如,在Ph+ 和低亚二倍体 ALL 中分别观察到与BCR::ABL1融合和 TP53 突变相关的CH(图2),这意味着应谨慎解释使用这些标志物进行的 MRD 监测,因为它们可能无法准确反映真实的残留原始细胞负荷。因此,IG/TR重排或继发性改变(例如IKZF1 缺失,如果在显性克隆中存在)是更可靠的 MRD 标志物。另一方面,其他亚型(如KMT2A-r)在整个患者病程中表现出特定的 IG/TR 重排克隆演变动力学,也可能影响 MRD 监测。
如果能够根据特异性标志物的异常表达模式识别并区分白血病细胞与正常前体 B 细胞,则也可以使用流式细胞术进行 MRD 监测,但该方法的适用性因基因亚型而异。例如,可免疫表型可能从 B 淋巴表型向髓系表型转换,特别是在通常显示谱系模糊并保留髓系潜力的基因亚型中,如KMT2A-r、ZNF384-r和DUX4-r。此外,B细胞靶向治疗可能掩盖 B 细胞表位,使残留白血病细胞的鉴定变得复杂。总之,B-ALL亚型内的生物学异质性对基于分子和流式细胞术的 MRD 方法都提出了挑战,需要高水平的专业知识。
B细胞靶向治疗时代的预后意义
随着 B 细胞靶向治疗的进步,B-ALL的治疗也在发生革命性变化,不同基因亚型的预后意义(目前主要与化疗敏感性相关)可能具有显著挑战性。药物反应与特定 B-ALL 亚型之间相关性的数据仍有限。CD19表面标记物是一种普遍存在的 B 细胞系标记物,在所有亚型中均有表达,可能不会影响CD19 靶向药物的初始反应。然而有多种治疗逃逸机制,如体细胞突变、剪接亚型选择、基因表达调节或谱系转换,这些都是抗原表位丢失的驱动因素。最近的数据表明,部分B-ALL亚型逃逸这些治疗的倾向更高。KMT2A-r和 ZNF384-r B-ALL 的髓系谱系较为混杂,通过转换为髓系细胞使 CD19 抗原损失的能力更高。低亚二倍体 ALL 可能更易受 CD19 基因损失的影响,因为其单等位基因状态与16号染色体复发性损失相关。特定异常可能与 B 细胞抗原表达差异有关,从而影响对这些药物的反应。例如,已报告 PAX5 P80R B-ALL 中 CD58 的下调会损害贝林妥欧单抗的有效性,因为 CD58 受 PAX5 正调控,与 CD2 相互作用增强 T 细胞应答。KMT2A-R B-ALL呈低CD22表面表达,可能导致药物偶联抗 CD22 抗体奥加伊妥珠单抗的反应较差。这些数据表明,B-ALL基因亚型(目前主要与化疗敏感性有关)相关的独特预后可能受到包含 B 细胞靶向药物的新治疗方案的挑战。对于化疗下预后良好的B-ALL亚型,考虑广泛降低化疗强度时需要谨慎。因此,全面的基因组分析和深入理解 B 细胞靶向药物反应的决定因素,对于建立未来 B-ALL 精准医学的基础都是必要的。
观点和挑战
尽管在 B-ALL中已经确定了几乎所有的复发性分子学改变,但在推进精准医疗方面仍然存在几个关键问题和挑战。首先,需要涉及大型患者队列的额外研究来巩固这些基因改变的临床意义,包括特定基因组合的影响,以及在 B 细胞靶向治疗背景下。实现这一目标可能需要大型国际合作研究。其次,更深入地了解这些改变驱动的致癌机制可以为创新的治疗策略铺平道路。最后,探索宿主因素在成人 B-ALL 启动和治疗应答中的作用,包括微环境和药物基因组学的影响,可能最终有助于开发优化的患者特异性治疗方法。
参考文献
Marie Passet, Rathana Kim, Emmanuelle Clappier; Genetic subtypes of B-cell acute lymphoblastic leukemia in adults. Blood 2024; blood.2023022919. doi: https://doi.org/10.1182/blood.2023022919
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言












#B-ALL# #基因亚型#
22