
罕见病专栏|成人期确诊STIM1基因突变所致Stormorken综合征1例
2025-03-27 中国神经精神疾病杂志 中国神经精神疾病杂志 发表于陕西省

该病多为儿童期起病,患者常就诊于儿童神经内科,现报道1例经我中心诊治成人期确诊的STIM1基因突变所致Stormorken综合征,并分析其临床、影像、病理及基因特点。
摘 要 报道1例成人期确诊的Stormorken综合征(Stormorken syndrome,STRMK),并研究其临床、病理及基因特点。患者为31岁男性,儿童期起病,以癫痫为首发症状,随后逐渐出现肌无力及肌萎缩。患者体格检查显示身材矮小,下肢近端肌力明显减退,伴有关节挛缩、脊柱侧弯。腹部CT检查提示脾脏缺如;双侧下肢肌肉MRI检查可见明显脂肪浸润。肌肉病理存在微管聚集(tubular-aggregate)现象。基因检测提示患者基质相互作用分子1(stromal interaction molecule 1, STIM1)基因存在c.326A>G(p.His109Arg) 杂合突变,为已知致病突变。通过此次病例报道使神经科医生进一步认识该罕见疾病。
关键词 Stormorken综合征;微管聚集性肌病;癫痫;基质相互作用分子;肌肉活检
Stormorken综合征(Stormorken syndrome,STRMK)是由编码参与钙库调控钙通道(stork operated calcium channel, SOC)的基质相互作用分子1(stromal interaction molecule 1, STIM1)基因突变所致[1-3]。可急性或慢性起病。急性起病者多以癫痫、惊厥或血小板减少所致皮肤黏膜出血为主要表现,而慢性起病则以肌病为主要表现[4-5]。该病多为儿童期起病,患者常就诊于儿童神经内科,现报道1例经我中心诊治成人期确诊的STIM1基因突变所致Stormorken综合征(Stormorken syndrome,STRMK),并分析其临床、影像、病理及基因特点。
1 临床资料
患者,男31岁,因“反复发作性意识丧失、肢体抽搐25年,四肢乏力伴肌肉萎缩16年”于2024年3月2日入住我院。患者25年前(6岁)无明显诱因突发意识丧失跌倒伴有四肢抽搐、双眼上翻,持续数分钟后缓解,外院就诊行头颅MRI检查未见明显异常,出院后予以“丙戊酸钠缓释片”治疗后,症状控制良好,随后3年未再发作并逐渐停药。患者21年前(9岁)再次出现反复意识丧失,伴有双眼愣神、呼之不应、大喊大叫、双手不自主摸索,每次持续1~3 min,每月发作10~20次,脑电图检查提示界限性脑电图、偶可见尖波;脑发育评估提示患者智力轻度低下、记忆力低下、适应性行为边界、注意力重度障碍;头颅MRI检查提示右侧海马异常信号影,诊断“部分性癫痫,简单部分性发作,复杂部分性发作,继发全面强直阵挛发作”,予以“丙戊酸钠缓释片、拉莫三嗪、卡马西平”联合治疗,患者症状部分好转,但仍有1~2月发作1次。患者自2008年起(15岁)逐渐出现四肢乏力,上坡抬腿费力,下蹲后站立困难,伴有肌肉萎缩,曾在当地就诊检查发现肌酸激酶水平升高、肌电图呈肌源性损害,长期予以康复治疗,近3年患者自觉四肢乏力及肌肉萎缩症状尚稳定,为进一步诊治入院。既往儿时有反复高热惊厥病史,家族史及个人史无特殊。入院后神经系统检查:身材矮小,身高151 cm,体质量39.3 kg,神志清楚,对答基本切题,双侧瞳孔等圆等大,直径2.5 mm,对光反射存在,其余脑神经检查未见明显异常。四肢肌肉容积均匀减少,四肢纤细,脊柱侧弯,双侧足趾关节挛缩(图1A),可见高弓足。双侧上肢肌力5级,双侧下肢近端肌力3级,远端背屈肌力5-级,跖屈肌力4级,肌张力正常,腱反射减退,病理反射未引出。双侧指鼻试验、跟膝胫试验稳准。感觉检查未见明显异常。行走鸭步,昂伯氏征阴性。蒙特利尔认知评估量表(Montreal cognitive assessment,MoCA)评分26分,简易智力状态检查量表(Mini-Mental State Examination,MMSE)评分30分。
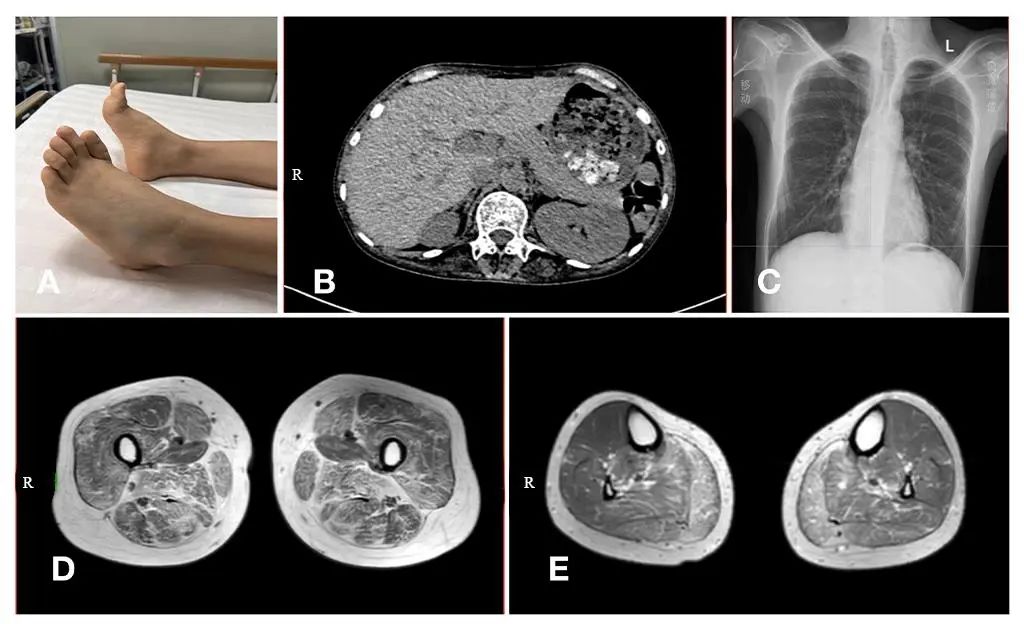
Fig.1 Images of the lower limbs and radiological examination in the patient with Stormorken syndrome图1 STRMT患者下肢图片及影像学检查结果 可见患者下肢足趾关节挛缩畸形(A);腹部CT检查提示脾脏缺如(B);胸部X线检查可见脊柱侧弯;双侧下肢MRI检查可见大腿前后肌群弥漫性脂肪浸润(D),双侧小腿以腓肠肌内侧头脂肪浸润明显(C)。
入院后实验室检查:肌酸激酶1519 U/L(30~310 U/L),乳酸0.98 mmol/L(0.7~2.1 mmol/L),谷丙转氨酶82 U/L(9~50 U/L)。腹部CT检查提示脾脏缺如(图1B)。胸部X线可见脊柱侧弯(图1C)。24 h视频脑电图示:醒-睡各期频繁可见右侧颞区大量高-极高波幅尖波、棘波、棘慢波、多棘慢波呈簇或连续发放;偶见左侧颞区高波幅尖波、尖慢波不同步发放。肌电图检查提示四肢骨骼肌肌源性损害。头部MRI和颅脑波谱分析(magnetic resonance spectroscopy MRS)提示:①双侧顶叶少许白质变性灶;②右侧海马较对侧稍小,双侧海马NAA峰及NAA/Cho+Cr值降低,右侧NAA/Cho+Cr值约为0.38~0.77,左侧NAA/Cho+Cr值约为0.33~0.93。双侧下肢肌肉MRI检查可见双侧臀大肌、大腿及小腿肌群弥漫性萎缩伴有脂肪变性(图1D和E)。心脏彩超未见明显异常。随后患者进一步行左侧腓肠肌活检,此外经患者及家属知情同意后进行外周血送第三方检测机构 (苏州赛福医学检验实验室) 行基因全外显子组测序。
光镜下苏木素-依红(hematoxylin and eosin, H-E)染色可见肌纤维大小不一,直径变异加大,少量结缔组织增生,未见明显肌纤维坏死再生。肌纤维内及肌膜下存在嗜碱性深紫色均匀细小颗粒状或絮状物质沉积,为微管聚集表现;经改良Gomori (modified Gomori trichrome, MGT)染色可见沉积物为紫红色,显示更为明显;还原型辅酶I四唑氮还原酶(nicotinamide adenine dinucleotide-tetrazolium reductase,NADH-TR)染色可见肌原纤维网格状结构紊乱,多数肌纤维可见酶活性缺失区,个别肌纤维见靶样或微小轴空结构;部分肌纤维胞质内存在NADH及过碘酸希夫染色(periodic acid-Schiff , PAS)酶活性明显升高表现;ATP9.4染色显示I型肌纤维占优(图2)。免疫组化结果提示肌纤维膜dystrophin、caveolin-3、dysferlin、dystroglycan染色未见异常。电镜检查提示部分肌原纤维灶性紊乱,肌小节消失及节段性坏死,可见个别少量管聚集结构,肌膜下线粒体增多,其中间可见空泡及类圆形物质(图2E)。
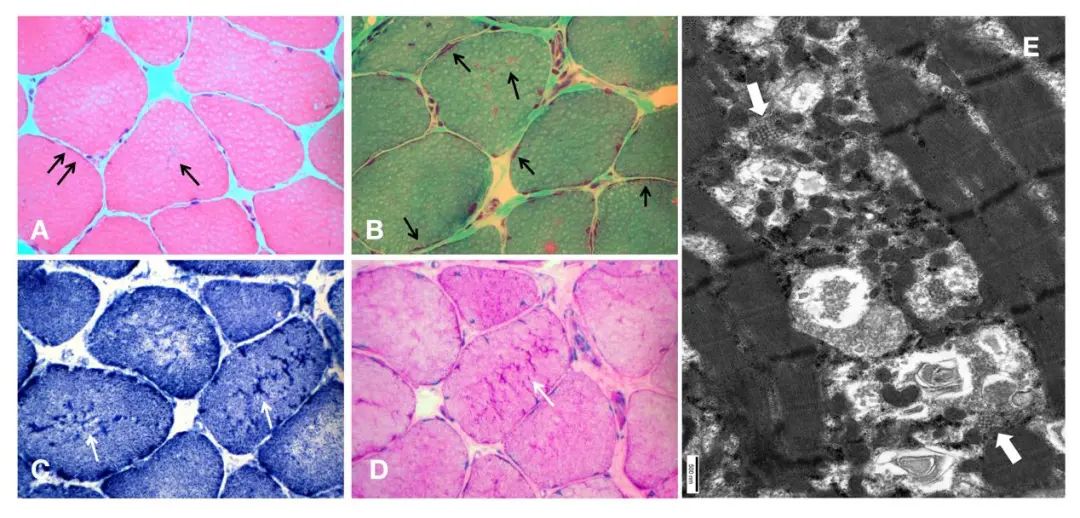
Fig.2 Histopathological examination results of the left gastrocnemius muscle in the patient with Stormorken syndrome图2 STRMT患者左侧腓肠肌肌肉病理检查结果 H-E染色可见肌纤维内及肌膜下存在嗜碱性深紫色均匀细小颗粒状或絮状物质沉积(A,×400);MGT染色可见沉积物为紫红色(B,×400);NADH-TR染色可见肌原纤维网格状结构紊乱, 部分肌纤维胞质内存在NADH-TR及PAS酶活性明显升高表现(C和D白色箭头,×400)。电镜检查提示部分肌原纤维灶性紊乱和少量管聚集结构(白色粗箭头)。
基因序列分析显示患者STIM1基因存在1个杂合突变c.326A>G(p.His109Arg) ,采用Sanger测序法对患者父母的STIM1基因变异进行验证,其父母均未发现该突变,提示该突变仅存在于患者,为新发杂合变异(图3A~C)。蛋白功能预测软件(SIFT、Polyphen_HDIV、Polyphen_HVAR、LRT、MutationTaster及MutationAssessor)提示此位点突变为致病位点,既往报道的文献中有8例患者和本例患者突变位点相同[3, 4, 6-8]。综合分析,该变异评级为致病性,结合患者的临床表现、辅助检查结果确诊为Stormorken综合征。患者出院至今口服丙戊酸钠、拉莫三嗪、卡马西平抗癫痫治疗,癫痫病情稳定,肌肉病变方面未见进展。
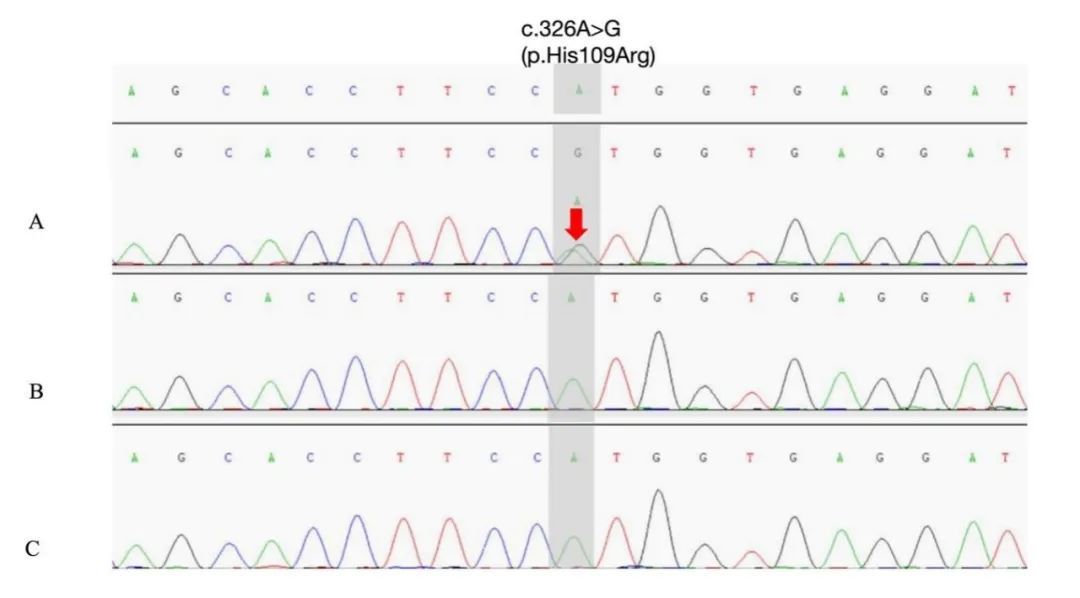
Fig.3 Sanger sequencing results of the STIM1 gene in the patient and their parents图3 患者及其父母STIM1基因Sanger测序结果 A.患者检测结果提示存在STIM1基因c.326A>G(p.p09R)杂合突变;B、C分别为患者父亲及母亲基因检测结果,无该位点突变。
2 讨论
微管聚集性肌病(tubular-aggregate myopathy, TMA)是一种罕见的遗传异质性较强的肌肉疾病,通常为散发或常染色体显性遗传,临床表现为肌无力、肌肉疼痛、痉挛和强直,下肢重于上肢,近端重于远端[9]。STRMK为一种罕见常染色体显性遗传疾病,最早在1983年被发现[7, 10],至目前为止国内仅有两例报道[4, 11],均为儿童期诊断。多数患者在儿童期发病,急性起病者多以癫痫、惊厥或血小板减少所致皮肤黏膜出血为主要表现,而慢性起病则以肌病为主要表现[4-5]。TAM是STRMK最主要的临床表型之一,患者常可见下肢近端无力合并肌肉萎缩,行走鸭步,部分患者合并有肌肉痉挛、疼痛、关节挛缩及肘关节过度松弛等,但也有少部分患者仅表现为肌酸激酶水平升高而无明显临床症状[5]。此外,STRMK患者可能伴有血小板减少、脾脏缺如、鱼鳞病、身材矮小、低钙血症及阅读障碍等多系统受累表现[4-5, 9-10, 12-13]。个别患者还可能出现心肌及呼吸肌受累,导致心功能不全及呼吸窘迫[8]。本例患者为青年男性,儿童期起病,临床表现为癫痫合并肌无力,体格检查可见患者身材矮小、关节挛缩、脊柱侧弯、无脾脏及下肢近端肌力减退,肌肉病理存在管聚集现象,STRMK临床异质性较强,当TAM合并多系统受累时则需重点考虑STRMK的可能性。
研究表明,肌肉MRI对于评估肌肉病变程度具有有一定价值。CLAEYS等[5]发现,STRMK患者病程越长,肌肉脂肪浸润越明显。全身肌肉MRI显示颈部伸肌、肩胛下肌、腹外斜肌、腰大肌均有受累;大腿前群肌肉以股直肌受累为主,后组肌群以股二头肌长头为主,小腿则以腓肠肌内侧头早期受累而胫前肌相对保留为特征改变。本研究中,患者下肢MRI显示腓肠肌内侧头脂肪浸润明显,与文献报道相一致;但大腿前组肌群中脂肪浸润相对平均,后组肌群则以大收肌和半膜肌受累为主,与文献报道不同。这可能与病程差异有关,提示STRMK患者受累肌群广泛,肌肉MRI特异性有限,仅作为辅助诊断手段。
肌肉病理检查是诊断STRMT的重要依据。STRMT患者肌肉活检显示I型及II型肌纤维胞浆内有嗜碱性异常物质沉积,MGT染色呈红色深染,NADH和PAS染色显示酶活性升高。电镜下,这些沉积物为规则排列的膜小管,称为管聚集物[7, 11, 14-15]。但管聚集物并非STRMT特有,也见于低钾性周期性麻痹、先天性肌无力综合征等[9]。免疫组织化学染色显示管聚集物内包含大量肌浆网蛋白,如STIM1和钙隔离素蛋白,因此推测管聚集物起源于肌浆网,因大量Ca2+内流引起肌浆网内错误折叠蛋白聚集,从而引起肌浆网结构功能破坏导致管聚集形成[16]。本例报道患者肌肉病理符合上述STRMT病理改变,但除典型管聚集外,该患者ATP染色可见I型肌纤维占优,NADH染色肌纤维内存在微小轴空样病变。上述两种病理改变也见于其他先天性肌病[17],提示SRTMK与其他先天性肌病在病理上有重叠,最终确诊需基因检测。
STRMK的主要致病基因为STIM1基因,此外还包括钙释放激活钙通道蛋白-1(calcium release-activated calcium channel protein 1,ORAI1)基因[8],两者均为编码SOCE的关键因子[6, 18]。STIM1基因在骨骼肌中高表达。其蛋白锚定在内质网/肌浆网上,其包含一个跨膜结构域,N端包含EF-手及SAM结构域,C端主要包含有3个线圈样结构域[2, 19]。本例患者的c.326A>G(p.His109Arg) 突变位点位于EF手结构域,为目前已知STIM1基因突变位点最常见的区域。该结构域主要位于内质网腔内,可结合钙离子,其突变可能导致蛋白质构象改变,引起EF手区域与SAM结构解离,导致钙离子内流和肌浆网功能破坏[9, 20]。目前我国已报道1例该位点突变的STRMK病例,其临床表现与本研究患者略有不同,既往报道起病年龄<3岁,以血肌酸激酶升高及血小板下降为主要临床特点[4, 14];因此该位点可能为我国患者的热点突变,但该病的基因型与临床表型之间相关性仍需要进一步研究。
3 点评
STRMK为一种常染色体显性遗传疾病,该病在临床上极为罕见,具有高度的临床异质性,当临床上出现微管聚集性肌病的临床表现同时合并多系统受累时需要考虑此综合征,基因检测对于早期诊断至关重要,目前,针对STIM1基因突变导致的STRMK综合征尚无特效治疗方法,但抑制钙信号传导途径和基因治疗等潜在治疗策略正在研究中,以期改善患者的临床预后。
致谢
感谢北京大学第一临床医院神经内科袁云教授在肌肉电镜病理检查中予以的帮助。
参考文献:
1. PECHE G A, SPIEGELHALTER C, SILVA-ROJAS R, et al. Functional analyses of STIM1 mutations reveal a common pathomechanism for tubular aggregate myopathy and Stormorken syndrome[J]. Neuropathology, 2020, 40(6): 559-569.
2. BÖHM J, LAPORTE J. Gain-of-function mutations in STIM1 and ORAI1 causing tubular aggregate myopathy and Stormorken syndrome[J]. Cell Calcium, 2018, 76: 1-9.
3. BÖHM J, CHEVESSIER F, MAUES DE PAULA A, et al. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy[J]. Am J Hum Genet, 2013, 92(2): 271-278.
4. 孙文强, 朱雪萍. STIM1基因突变导致Stormorken综合征临床报道并文献复习[J]. 国际儿科学杂志, 2022, 11(49): 782-785.
5. CLAEYS T, GOOSENS V, RACÉ V, et al. Clinical and muscle MRI features in a family with tubular aggregate myopathy and novel STIM1 mutation[J]. Neuromuscul Disord, 2020, 30(9): 709-718.
6. NOURY J B, BÖHM J, PECHE G A, et al. Tubular aggregate myopathy with features of Stormorken disease due to a new STIM1 mutation[J]. Neuromuscul Disord, 2017, 27(1): 78-82.
7. LI A, KANG X, EDELMAN F, et al. Stormorken Syndrome: A Rare Cause of Myopathy With Tubular Aggregates and Dystrophic Features[J]. J Child Neurol, 2019, 34(6): 321-324.
8. TICCI C, CASSANDRINI D, RUBEGNI A, et al. Expanding the clinical and genetic spectrum of pathogenic variants in STIM1[J]. Muscle Nerve, 2021, 64(5): 567-575.
9. MORIN G, BIANCALANA V, ECHANIZ-LAGUNA A, et al. Tubular aggregate myopathy and Stormorken syndrome: Mutation spectrum and genotype/phenotype correlation[J]. Hum Mutat, 2020, 41(1): 17-37.
10. STORMORKEN H, SJAASTAD O, LANGSLET A, et al. A new syndrome: thrombocytopathia, muscle fatigue, asplenia, miosis, migraine, dyslexia and ichthyosis[J]. Clin Genet, 1985, 28(5): 367-374.
11. JIANG L J, ZHAO X, DOU Z Y, et al. Stormorken Syndrome Caused by a Novel STIM1 Mutation: A Case Report[J]. Front Neurol, 2021, 12: 522513.
12. DE LA FUENTE-MUNOZ E, VAN DEN RYM A, GARCÍA-SOLIS B, et al. Case Report: Novel STIM1 Gain-of-Function Mutation in a Patient With TAM/STRMK and Immunological Involvement[J]. Front Immunol, 2022, 13: 917601.
13. SURA A, JACHER J, NEIL E, et al. Chronic Thrombocytopenia as the Initial Manifestation of STIM1-Related Disorders[J]. Pediatrics, 2020, 145(4):e20192081.
14. SUN W, HU J, LI M, et al. Stormorken syndrome caused by STIM1 mutation: A case report and literature review[J]. Med Int (Lond), 2022, 2(5): 29.
15. BORSANI O, PIGA D, COSTA S, et al. Stormorken Syndrome Caused by a p.R304W STIM1 Mutation: The First Italian Patient and a Review of the Literature[J]. Front Neurol, 2018, 9: 859.
16. FAN Q, GWATHMEY K, DU X, et al. Tubular aggregate myopathy causing progressive fatiguable weakness[J]. Pract Neurol, 2024, 24(2): 137-140.
17. FUSTO A, CASSANDRINI D, FIORILLO C, et al. Expanding the clinical-pathological and genetic spectrum of RYR1-related congenital myopathies with cores and minicores: an Italian population study[J]. Acta Neuropathol Commun, 2022, 10(1): 54.
18. SILVA-ROJAS R, PÉREZ-GUÀRDIA L, SIMON A, et al. ORAI1 inhibition as an efficient preclinical therapy for tubular aggregate myopathy and Stormorken syndrome[J]. JCI Insight, 2024, 9(6): 1-17.
19. DARBELLAY B, ARNAUDEAU S, BADER C R, et al. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release[J]. J Cell Biol, 2011, 194(2): 335-346.
20. STATHOPULOS P B, ZHENG L, LI G Y, et al. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry1 [J]. Cell, 2008, 135(1): 110-122.
【引用格式】刘丽丽,朱琳,胡俊,等. 成人期确诊STIM1基因突变所致Stormorken综合征1例[J]. 中国神经精神疾病杂志,2025,51(1):60-64.
【Cite this article】LIU L,ZHU L,HU J,et al.A comprehensive report on the clinical, pathological, and genetic characteristics of Stormorken syndrome attributed to a STIM1 gene mutation, diagnosed in adulthood[J]. Chin J Nervous Mental Dis,2025,51(1):60-64.
DOI:10.3969/j.issn.1002-0152.2025.01.011

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





#Stormorken综合征# #微管聚集性肌病#
6