
【衡道丨干货】2025 中国肿瘤整合诊治指南(CACA) ——淋巴瘤
2025-03-26 衡道病理 衡道病理 发表于陕西省

《2025 中国肿瘤整合诊治指南(CACA)——淋巴瘤》详细介绍了淋巴瘤的诊疗总则、病理分类、活检与制片、组织病理学检查、流式细胞术分析、遗传学与分子病理检测等内容。
《2025 中国肿瘤整合诊治指南(CACA)——淋巴瘤》详细介绍了淋巴瘤的诊疗总则、病理分类、活检与制片、组织病理学检查、流式细胞术分析、遗传学与分子病理检测等内容。指南强调了淋巴瘤的异质性,提出了基于组织形态、免疫组化、分子遗传学等多方面的综合诊断方法。此外,指南还涉及了弥漫大B细胞淋巴瘤、高级别B细胞淋巴瘤、原发纵隔大B细胞淋巴瘤、原发中枢神经系统淋巴瘤、其他结外淋巴瘤等特定类型的淋巴瘤的病理诊断。
淋巴瘤的诊疗总则
概述
淋巴瘤是起源于淋巴结和淋巴组织的恶性肿瘤,其发病率逐年上升,严重威胁人类的健康。淋巴瘤病理类型复杂,异质性强,治疗策略和预后各不相同。临床上主要表现为无痛性淋巴结肿大,肝脾肿大,全身各组织器官均可受累,可伴发热、盗汗、消瘦、瘙痒等全身症状。根据病理类型不同分为非霍奇金淋巴瘤(NHL)和霍奇金淋巴瘤(HL)两类。NHL发病率远高于HL,是具有很强异质性的一组疾病。淋巴瘤的病因尚不清楚,一般认为,可能和病毒等微生物感染、放射线、化学药物、自身免疫病、基因突变等有关。我国淋巴瘤的发病率约为6.68/10万,且以每年3%~5%的比例增长,目前每年大约有10万例新发淋巴瘤患者,已成为我国男性前十大高发恶性肿瘤。
病理分类
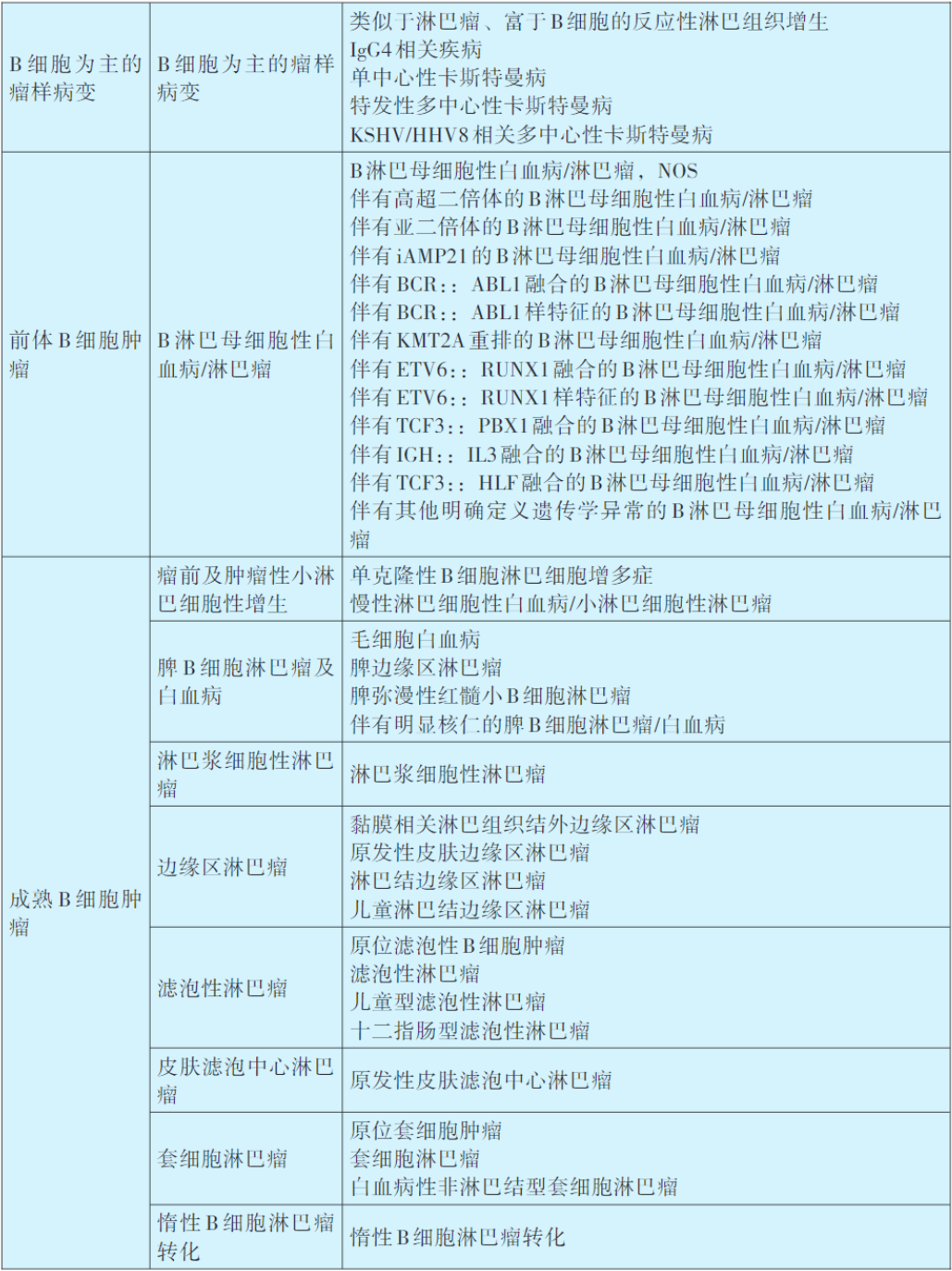
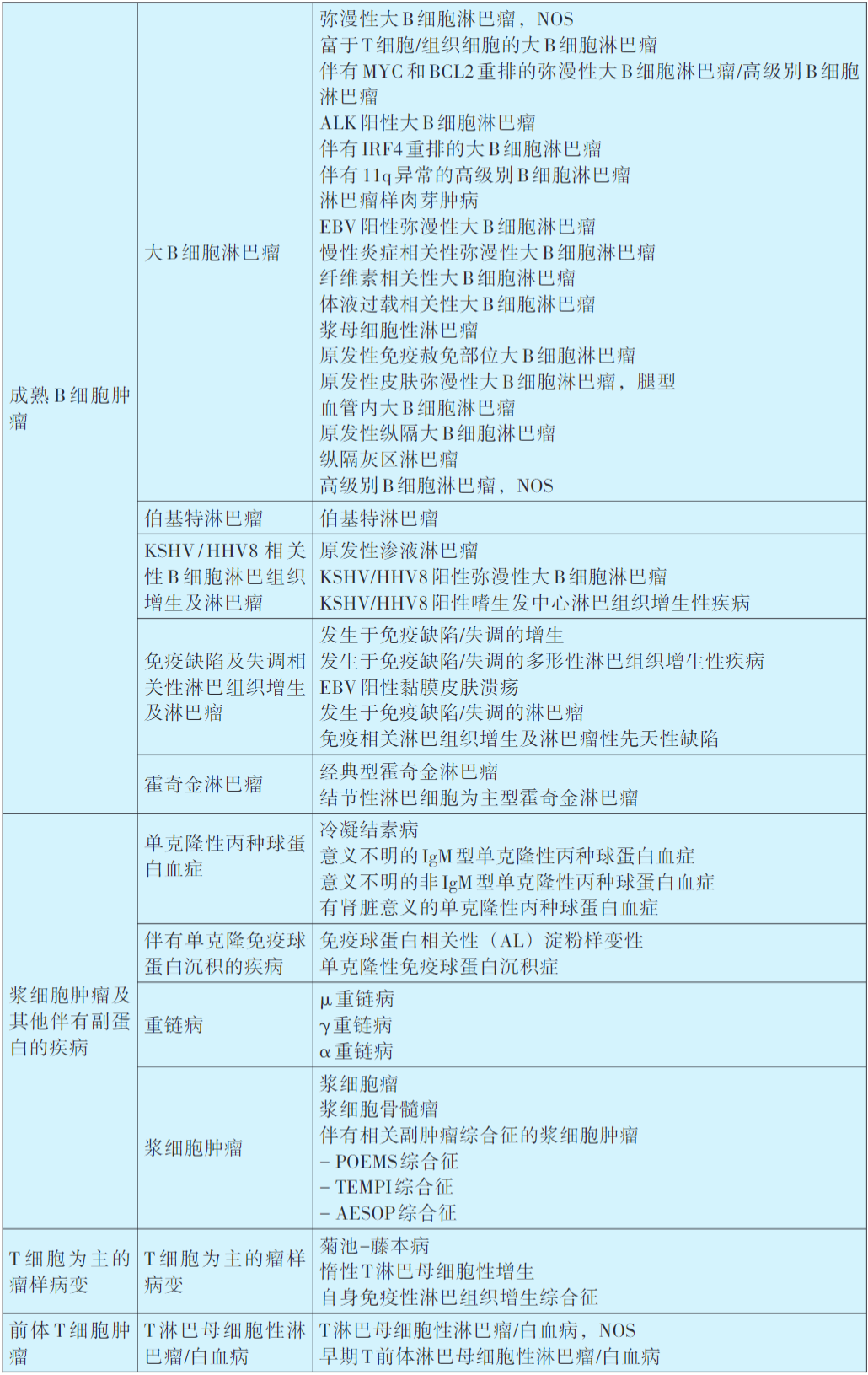
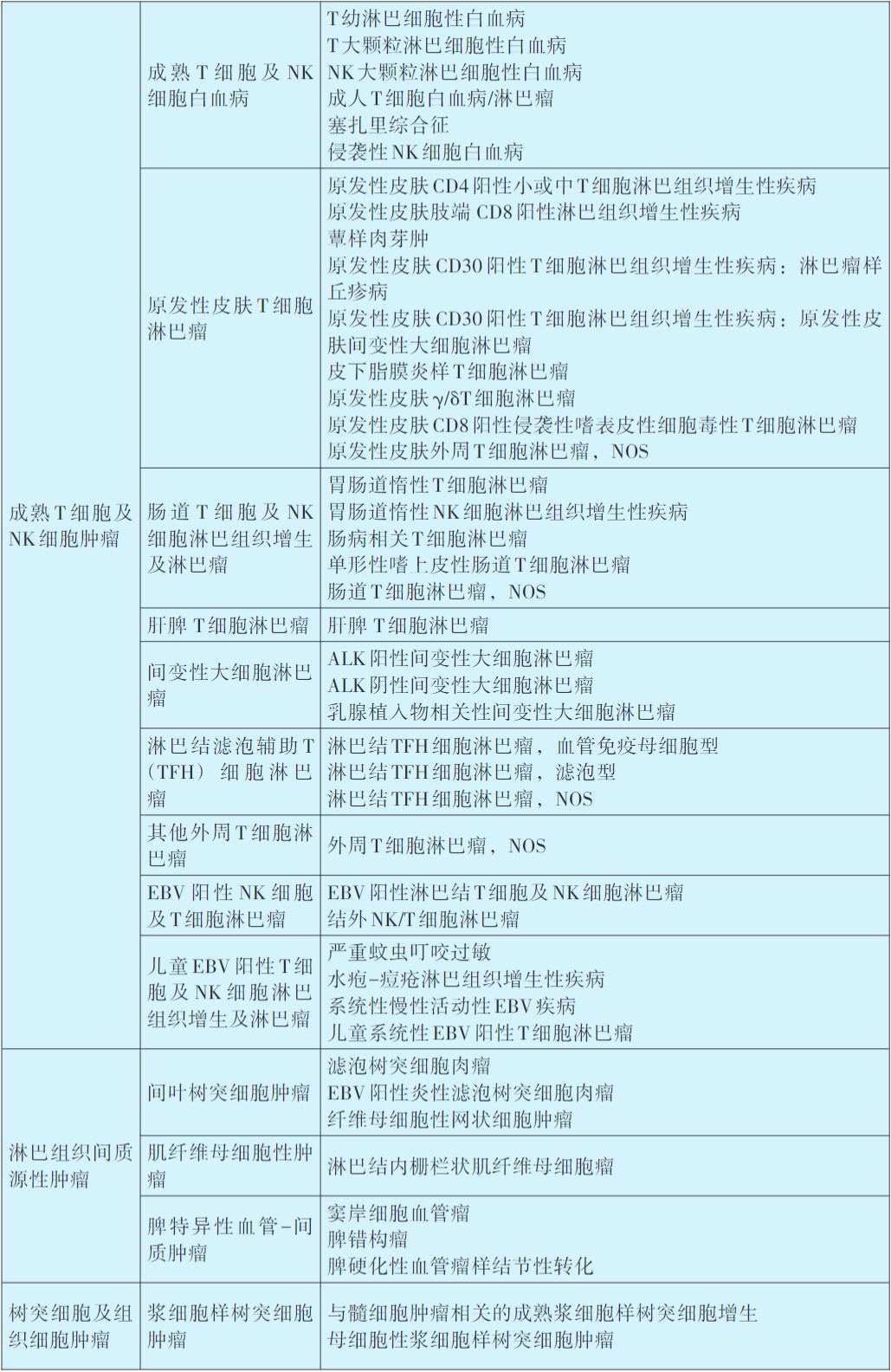
淋巴瘤WHO分类及诊断原则
目前,淋巴瘤的类型区分和诊断标准主要是依据世界卫生组织(WHO)制订的造血和淋巴组织肿瘤分类(详见下表)。淋巴瘤病理诊断整合了组织形态、免疫组织化学染色、流式细胞分析、细胞遗传学以及分子生物学等多种辅助检测技术。迄今为止,组织病理学检查仍然是绝大部分淋巴瘤病例的确诊方法,而免疫组织化学染色则是判断肿瘤免疫表型以及检测部分遗传学异常的重要手段。所以,几乎所有淋巴瘤病例均需接受包括免疫组化在内的组织病理学检查之后方能确诊,部分病例的诊断和鉴别,还需辅以其他必要的检测技术。
独特的临床特点也是某些类型淋巴瘤确诊的重要依据,申请病理检查的临床医师有义务通过填写病理检查申请单提供必要的信息(包括患者的年龄、性别、活检部位等一般信息以及临床表现、影像学、内镜和其他实验室检查的主要阳性发现、既往诊断、治疗史等)。病理医师也可通过查阅电子病历、直接与临床医师沟通或参加多学科整合诊疗(MDT to HIM)讨论等多种形式获得相关信息
2016年修订第4版WHO造血和淋巴组织肿瘤分类

活检与制片
标本获得
淋巴瘤首次病理诊断必须根据切除或切取活检所获得的组织标本做出。足量、合格的诊断性组织是对淋巴瘤进行形态观察以及开展免疫表型和遗传学研究的基础,必要时应重复活检。淋巴结或某些结外病灶的完整切除标本,有助于病理医师对整个病变进行全面评估,且有足量的组织用于辅助检查,是诊断淋巴瘤最为理想的标本。如有多个解剖区域的淋巴结病灶,一般宜选择颈部病灶。对难以完整切除的病灶,可通过开放手术、内镜下活检或空芯针穿刺等方法获得小块组织样本供病理学检查,多数也能满足诊断需要。一般而言,细针吸取细胞学检查不能作为淋巴瘤的首诊依据,但可用于淋巴瘤疑似病例的初筛以及部分确诊病例可疑或复发病灶的确认,在某些特定情形下(例如:非实体性淋巴肿瘤、体液标本或获得病变组织较为困难时),细胞学检查亦可用于疾病诊断,但常需辅以细胞块制作、免疫组化、流式细胞或细胞遗传学分析等辅助检查。
组织处理
原则上,所有淋巴结或体积较大的淋巴瘤组织标本均应在新鲜、湿润状态下尽快送到病理科处理,不能及时送检的标本可用生理盐水湿纱布包裹后放置4℃冰箱短暂保存。病理科在接收标本后应尽快处理。较大的淋巴结标本应垂直其长轴做平行切分(每片组织厚度0.3~0.5cm),小于1cm的淋巴结可沿淋巴结长轴最大面对剖。可先行快速病理检查(冷冻切片或印片)以初判是否淋巴造血组织肿瘤,对疑似淋巴瘤的病例,应选择1~2片最大的组织标本浸于4%中性甲醛溶液固定,固定时间常为12~24小时,及时和适当时间的固定是制作高质量淋巴瘤组织切片的重要前提。剩余组织可分别用于生物样本库存档、流式细胞分析、细胞遗传学检查、病原微生物检测等。对非淋巴瘤或疑似感染性病变的标本,应尽快将所有组织固定。对体积较小的切取、钳取或穿刺活检标本,应先固定,然后再送病理科检查。对骨髓活检标本,还应在固定后进行脱钙处理。标本组织固定后还需脱水、透明、浸蜡、包埋等程序化加工才能制作切片,上述组织处理步骤目前多在自动组织处理仪中完成。
切片制作
高质量的常规苏木精-伊红(HE)染色切片是淋巴瘤病理诊断的重要依据。HE染色切片质量优劣与否,取决于组织处理、切片、染色、封固等诸多技术环节的质量控制。其中,及时而充分的固定、浸蜡前彻底脱水以及封片前透明这些步骤尤为关键,切片厚度以2~4μm为宜。概括而言,一张高质量的切片,应该达到组织固定良好、组织平整、无刀痕或气泡、染色鲜艳、组织及细胞结构清晰、封固良好等技术要求
术中冷冻切片检查对初步区分淋巴瘤与非淋巴造血组织肿瘤有一定价值,但常不足以确诊淋巴瘤。淋巴瘤印片检查是组织切片检查的有益补充,以其方法简便、操作快捷而常被用于淋巴瘤的快速筛查。
组织病理学检查
组织学形态分析
基于常规HE染色切片的组织形态分析尤为重要。一方面,特征性的形态改变本身就对某些类型淋巴瘤的诊断有着决定性提示作用;另一方面,相当多的辅助检查(例如:免疫表型分析、分子遗传学检测等)都必须在形态分析的基础上合理选择和使用。概括而言,淋巴瘤组织形态分析的基本原则和其他实体肿瘤相似,恶性肿瘤的一些共同特性,例如瘤细胞的异型性和破坏性生长等,在各种淋巴瘤中也有相应的表现,且常是淋巴瘤和反应性病变鉴别的重要依据。需要指出的是,淋巴瘤的形态分析通常离不开免疫组化染色的帮助。
免疫组化检查
(1)免疫组化的作用
免疫组化检查对淋巴瘤诊断与鉴别诊断的作用主要体现在以下几个方面:①判断肿瘤的细胞系(例如:B细胞或T、NK细胞淋巴瘤);②判断肿瘤性免疫细胞的分化阶段和成熟程度(例如:淋巴母细胞淋巴瘤与外周B/T细胞淋巴瘤、滤泡性淋巴瘤与边缘区淋巴瘤等);③检测某些遗传学改变(例如:CCND1、ALK等基因易位所导致的蛋白异常表达);④鉴别良、恶性疾病(例如:通过检测免疫球蛋白轻链有否限制性表达来判断B细胞/浆细胞是否克隆性增生);⑤检测病原微生物(例如:EBV、HHV8、幽门螺杆菌等);⑥为临床免疫或靶向治疗提供依据(例如:CD20、CD30、CD19、CD38、PD-L1、ALK等靶点的检测);⑦提示疾病预后(例如:通过检测CD10、BCL6、MUM1等指标来区分弥漫性大B细胞淋巴瘤的COO分型;通过检测MYC与BCL2蛋白表达水平来甄别“双表达”淋巴瘤)。
(2)常用标志物
可用于淋巴瘤石蜡包埋组织免疫染色的常用标志物包括以下几大类:①白细胞共同抗原(CD45/LCA);②B细胞相关标记物,例如CD20、CD79a、CD19、PAX5、0ct-2、BOB.1、κ、λ、IgG、IgG4、IgM、IgA、IgD、CD38、CD138、CD23等;③T细胞/NK细胞相关标记物,例如CD3、CD2、CD5、CD7、CD4、CD8、CD43、CD45RO、CD56、CD57、细胞毒性分子(包括TIA-1、颗粒酶B、穿孔素)、T细胞受体蛋白(例如βF1、TCRG)等;④淋巴细胞活化/分化相关标记物,例如CD30、TdT、CD99、CD10、BCL6、MUM1等;⑤肿瘤基因和增殖相关标记物,例如ALK、BCL2、BCL10、cyclin D1、MYC、TP53、Ki-67等;⑥组织细胞、树突细胞及髓系相关标记物,例如CD68(KP1、PGM1)、CD163、溶菌酶、髓过氧化物酶(MPO)、CD15、CD123、CD117、CD21、CD35、S-100、CD1a、CD207/langerin等;⑦微生物标志物,例如EB病毒(EBV)-LMP1、HHV8等;其他,例如EMA、细胞角蛋白、LEF1、MNDA、PD1、PD-L1、CXCL13等
(3)免疫组化诊断注意事项
①免疫组化检查首先应确保染色质量,一定要从组织处理、制片、抗原修复、抗体选择、染色程序等诸多环节加强监控,并通过设置合理的阳性对照作平行染色,以确保染色质量稳定保持在较高水平。②要熟悉各类淋巴瘤组织学形态和免疫表型,在形态分析基础上,有所针对地选择必要的抗体组合来证实诊断或帮助鉴别,不应使用抗体“大套餐”作过度检测。③应学会正确判读免疫组化染色结果。这就要求病理医师做到:(a)熟悉各种抗体的预期染色结果,并通过适当内、外对照来判断染色成功与否;(b)在形态分析基础上正确判断何种细胞成分表达何种抗原;(c)悉各种抗体的反应谱系和适用范围,避免片面或错误解读阳性结果。
(4)常用标志物组合的选择
①对需做免疫组化的淋巴组织增生性病变,几乎所有病例都需要检测CD20、CD3和Ki-67。这一组合能够突显淋巴组织的免疫结构,有助于良、恶性病变的鉴别,并能提示淋巴瘤的细胞系起源;②对呈滤泡/结节状生长模式的病变,可选择CD10、BCL6、CD21、Ki-67等指标来显示结节和淋巴滤泡的关系;③对疑似小B细胞肿瘤性病变(包括低级别滤泡性淋巴瘤、慢性淋巴细胞性白血病/小淋巴细胞性淋巴瘤、套细胞淋巴瘤、边缘区淋巴瘤等),可选用CD10、BCL6、CD5、CD23、cyclin D1、SOX11、LEF1和MNDA这一组指标予以鉴别诊断;④对富含浆细胞的病变,可检测免疫球蛋白轻链(κ/λ)有无限制性表达以区分良、恶性;⑤对疑似高侵袭性成熟B细胞肿瘤的病变[包括绝大部分弥漫性大B细胞淋巴瘤、伯基特淋巴瘤以及具有前二者中间特征的B细胞淋巴瘤(BCLU)或高级别B细胞淋巴瘤(HGBL)、高级别滤泡性淋巴瘤等],选用CD10、BCL6、BCL2、MUM1、MYC这一组指标(并结合细胞遗传学检查)有助确诊并区分亚型;EBV-LMP1、CD5和TP53的检测对于弥漫性大B细胞淋巴瘤有预后意义;⑥对疑似T细胞或NK细胞肿瘤的病变,可选择性检测CD2、CD5、CD7、CD4、CD8、CD10、CD30、CD56、ALK、CXCL13、PD1、T细胞受体蛋白、细胞毒性分子等标志物并行EBER原位杂交来帮助判断肿瘤类型;⑦对经典型霍奇金淋巴瘤或类似病变(例如:具有经典型霍奇金淋巴瘤和弥漫性大B细胞淋巴瘤中间特征的灰区淋巴瘤、结节性淋巴细胞为主型霍奇金淋巴瘤、富于T细胞/组织细胞的大B细胞淋巴瘤等),可选用CD20、PAX5、0ct-2、BOB.1、CD30、CD15、EBV-LMP1(或EBER)、EMA、IgD、PD1等指标组合,此外,还应注意部分外周T细胞淋巴瘤也可伴有霍奇金样异型大B细胞浸润,增生的T细胞有无异型性、是否克隆性增生是鉴别诊断的关键;⑧富于细胞的经典型霍奇金淋巴瘤与ALK阴性的间变性大细胞淋巴瘤有时不易区分,检测B、T细胞系标志物、细胞毒分子并结合IG、TCR基因重排检测会有帮助。⑨对混合B、T细胞增生性病变,应结合形态分析正确区分肿瘤细胞和反应性成分。少数情况下,也不排除组合表型的淋巴瘤可能,但诊断后者应有充分的病理学和分子遗传学证据;对形态高度疑似淋巴造血组织肿瘤、但CD20和CD3均不表达的病变,通常需要检测部分“二线”细胞系标志物(例如:CD79a、PAX5、CD19、0ct-2、BOB.1、浆细胞相关抗原、CD3以外的全T细胞抗原以及CD43、CD68、MPO等髓细胞标志物等)帮助判别细胞系。
流式细胞术分析
基于流式细胞技术的免疫表型分析也是淋巴瘤诊断和分型的重要手段,有技术条件的病理实验室应积极开展。相比免疫组化,流式细胞术具有敏感度高、特异性强、检测周期短等特点,特别是对判断B、T细胞的克隆性增生、抗原表达水平以及小B细胞类肿瘤鉴别诊断等方面具有独特优势,弱点在于不能结合组织学形态分析(免疫组化可以在原位标记抗原)、不适合检测部分定位于细胞核或细胞浆内的抗原(例如:BCL6、MUM1、cyclin D1、Ki-67、BCL2等)、对霍奇金淋巴瘤等肿瘤细胞较少的病变以及T细胞或NK细胞肿瘤的甄别能力不如免疫组化强。此外,流式细胞分析需要细胞悬液或由新鲜组织制备的单细胞悬液标本,不常规留用新鲜组织标本的单位无法开展这项技术,细胞悬液标本也不像组织块那样可以长期保存,故而流式细胞术不能用于回顾性研究。
遗传学与分子病理检测
淋巴瘤中抗原受体基因(IG、TCR)的克隆性基因重排、非随机、类型相关性染色体及基因异常、特定病原微生物感染等不仅对研究肿瘤的发生、发展机制具有重要意义,也是精确诊断疾病、指导规范治疗及预测预后必不可少的工具。常用淋巴瘤遗传与分子病理检测方法包括聚合酶链反应(PCR,包括RT-PCR、RQ-PCR等)和Sanger测序技术、荧光原位杂交(FISH)、原位杂交(ISH)、核型分析(包括G显带、M-FISH、SKY等)以及基因表达谱(GEP)、二代测序(NGS)等高通量检测技术。
克隆性IG和TCR 基因重排检测
(1)方法
多数实验室采用PCR法并应用BIOMED-2引物组检测,以毛细管电泳基因扫描分析结果(或PAGE电泳异源双链分析)。
(2)适用范围
绝大部分淋巴组织增生性病变根据形态特征并结合免疫组化检查和临床特点便能确诊,无需开展这项检测。仅在少数情形下,克隆性IG和TCR基因重排检测对于淋巴瘤的诊断与鉴别、瘤细胞系确定以及克隆相关性分析具有一定价值:①良、恶性较难鉴别的病变,例如,淋巴瘤局限或隐匿性累犯、形态异常不显著或缺乏特征性免疫表型的淋巴瘤(例如:在某些炎性疾病基础上发生瘤变的早期MALT型边缘区淋巴瘤、EBV相关淋巴瘤等)、小细胞性皮肤淋巴瘤早期病变等;②疑似淋巴瘤、但标本组织较小较少,例如,不理想的穿刺活检或内镜活检标本、体液标本等;③某些特定病种的诊断与鉴别,例如,儿童型滤泡性淋巴瘤、淋巴瘤样丘疹病、水疱-痘疮样淋巴瘤等;④细胞构成较复杂或免疫标记难以区分细胞系的肿瘤,例如,肿瘤细胞异常表达CD20的外周T细胞淋巴瘤、伴有B细胞成分旺炽增生的外周T细胞淋巴瘤或B、T细胞组合性淋巴瘤等;⑤肿瘤克隆相关性分析,例如,判断弥漫性大B细胞淋巴瘤是否由之前滤泡性淋巴瘤转化而来;⑥微小残留病灶评估。
(3)判读结果注意事项
IG和TCR基因克隆性重排检测结果,一定要在组织病理学检查的背景下解读才有意义,如与形态或免疫组化证据不符,一般更倾向于组织学检查结论。判读基因重排结果,应注意以下事项:①克隆性增生不一定等于淋巴瘤,部分良性病变也可有淋巴细胞克隆性增生;②部分B或T细胞淋巴瘤(特别是淋巴母细胞性肿瘤、血管免疫母细胞性T细胞淋巴瘤等)IG和TCR基因重排检测结果存在谱系交叉,不足以判断瘤细胞系起源,此外,TCRB和TCRG基因重排也并不代表就是αβ和γδT细胞来源的肿瘤;③由于PCR技术的高敏性,标本组织中较少的细胞成分有时会产生假克隆或寡克隆,需与真性克隆性病变鉴别。④某些技术因素也会导致假阳性或假阴性结果
FISH法检测非随机性染色体和基因异常
部分B细胞非霍奇金淋巴瘤亚型和少数T细胞淋巴瘤具有特征性的、非随机性染色体异常(例如:染色体易位、缺失等),并导致相关基因异常,检测这些遗传学异常,有助于病理诊断或预后评估。目前,FISH是临床检测这些染色体/基因异常最常用的方法,也有多种针对染色体易位断裂区和基因缺失(或扩增)的商品化探针供应,针对易位的探针又包括融合探针和分离探针两种,分别是针对不同基因或同基因断裂位点两侧序列而设计,前者例如t(14;18)(IgH/BCL2)、t(11;14)(IgH/CCND1)等,后者例如t(18q21)(BCL2)、t(3q27)(BCL6)、t(8q24)(MYC)、t(14q32)(IgH)、t(18q21.31)/MALT1等。需要指出的是,部分染色体易位/基因重排可通过更为简易、经济的免疫组化法予以间接提示,例如,套细胞淋巴瘤相关的t(11;14)和间变性大细胞淋巴瘤相关的t(2p23)就分别可以通过cyclin D1和ALK的免疫组化染色来加以显示,在这些情形下,FISH检测就并非必需。但对那些蛋白表达并不一定对应于基因异常的情形(例如:弥漫性大B细胞淋巴瘤中BCL2和/或BCL6与MYC基因重排检测、有BCL2基因易位但免疫组化结果阴性的滤泡性淋巴瘤等),FISH检测就是必要方法。此外,部分遗传学异常对应于肿瘤的生物学异质性,例如,伴有t(2p23)(ALK)、t(6p25)(DUSP22-IRF4)和t(3q28)(TP63)的间变性大细胞淋巴瘤以及伴有del(17p)、del(11q)、del(13q)、+12等异常的慢性淋巴细胞性白血病/小淋巴细胞性淋巴瘤就有着不同的生物学行为,通过FISH检测这些遗传学异常,能提示疾病预后,并指导治疗。
EBER原位杂交检测
EBV感染与多种良、恶性淋巴组织增生性疾病相关。EBER-1/2是EBV编码的两个小分子量早期核糖核酸,常高水平地表达于病毒感染的细胞核中。利用EBER探针作原位杂交可以敏感地在原位显示病毒感染,如结合细胞系标志物免疫染色作双重标记,还能显示病毒阳性细胞的表型。通过免疫组化检测EBV编码的部分蛋白抗原(例如:LMP1、LMP2A、EBNA等)虽也能显示病毒存在,但这些抗原的表达情况在病毒不同感染模式中有所不同(例如:EBV阳性的经典型霍奇金淋巴瘤通常表达LMP1,而EBV阳性的伯基特淋巴瘤则通常LMP1阴性),而EBER却是恒定表达的。且免疫组化检测灵敏度也往往不如原位杂交,因此,EBER原位杂交技术常被视作组织内原位检测EBV的“金标准”
二代测序、基因表达谱等高通量技术检测
随着分子生物学研究深入,一些重现性基因突变(或其他异常)被发现在特定类型的淋巴瘤中高频发生,提示这些异常可能参与了肿瘤的发生、发展机制,其中有不少特定的基因突变已被应用于淋巴瘤的诊断分型、预测预后,乃至辅助临床作治疗决策。近年来,Sanger测序、二代测序等技术被越来越多用到淋巴瘤的分子病理诊断中,特别是高通量的二代测序技术具有单次实验能够检测多个基因变化以及多种遗传学异常(基因突变、易位、缺失等)的优势,大有替代其他测序技术的趋势。就淋巴瘤相关基因二代测序在临床应用而言,建议优先选择一组与诊断、预后判断和治疗选择密切相关的基因进行检测。基因表达谱是指一次同时定量检测特定组织中成千上万个基因的表达,再根据基因表达种类和丰度信息,构建出基因表达的数据表或谱型(或称指纹)。在淋巴瘤领域,弥漫性大B细胞淋巴瘤是第一种通过基因表达谱信息进行分子分型的肿瘤。此外,nCounter技术也能高度灵敏地定量检测多种样品类型(纯化总RNA、细胞和组织裂解液、石蜡包埋组织提取的RNA等)中的基因表达,该技术应用分子条形码和单分子成像来检测并计数单个反应中的几百个转录本,而不需要逆转录或扩增反应,直接数字化读出每一种mRNA的相对丰度。利用Nanosting平台的20基因检测(Lymph2Cx)研究已表明该项技术可对弥漫性大B细胞淋巴瘤石蜡包埋标本进行准确的分子分型。
弥漫大B细胞淋巴瘤
概述
弥漫大B细胞淋巴瘤(diffuse Large B-cell Lymphoma,DLBCL)是NHL中最常见的类型,在西方国家约占NHL的30%~40%,在我国占比更高,约占NHL的50%。中位发病年龄为50~70岁,男性略高于女性。多数为原发,也可由惰性淋巴瘤转化而来。根据细胞起源,DLBCL分为生发中心型和非生发中心型。R-CHOP为基础的一线治疗方案,约60%的患者可达治愈,但仍有30%~40%的患者发展为复发难治,近年新的免疫靶向治疗药物有望改善患者的预后。
病理诊断
诊断DLBCL常规IHC标志物包括CD19、CD20、PAX5、CD3、CD5、CD79α、Cyclin D1、Ki-67;常表现为CD19(+)、CD20(+)、PAX5(+)、CD3(-)。通过检测基因表达谱,根据细胞起源(cell of origin,COO)的不同将DLBCL分为3类,即生发中心B细胞样(germinal center B-cell like,GCB)型、活化B细胞样型(activated B-cell like,ABC)和第3型。临床上常用Han's分型进行分类,分为GCB型及非生发中心B细胞样(non-germinal center B-cell like,non-GCB)型,其中GCB型的IHC表现为:①CD10(+)、不论BCL-6和MUM1表达如何;②CD10(-)、BCL-6(+)、MUM1(-);其他情况均为non-GCB型。
2022年第5版 WHO淋巴造血肿瘤分类将伴MYC和BCL2基因易位,即遗传学特征为同时存在MYC和BC12基因重排的DLBCL,列为一个独特分类,称为“双打击”DLBCL,也称为高级别B细胞淋巴瘤。“双表达”DLBCL,即MYC蛋白表达>40%,BCL2蛋白表达>50%。“双表达”DLBCL往往提示预后不良。
高级别B细胞淋巴瘤
概述
高级别B细胞淋巴瘤(high grade B cell lymphoma,HGBL)是一种形态学和遗传学特点介于DLBCL和伯基特淋巴瘤(Burkitt'slymphoma,BL)之间的高度侵袭性淋巴瘤。2022年第5版WH0分类将HGBL分为:①伴有MYC和BCL2重排(不伴或伴有BCL6基因重排,即所谓“双打击”或“三打击”)的HGBL(HGBL-MYC/BCL2):②伴有11q染色体异常的HGBL(HGBL-11q,伴有11q获得/缺失,形态、表型及基因表达谱类似于BL或其他HGBL,但无MYC重排,且基因突变特征不同于BL);③除此之外的HGBL归为非特指型(HGBL-NOS)。仅伴有MYC和BCL6重排(但没有BCL2重排)的双打击淋巴瘤在新分类中不再归入伴有MYC和BCL6重排的HGBL,而是归入HGBL-NOS或DLBCL-NOS。HGBL发病率仅占非霍奇金淋巴瘤的1%~2%。
病理诊断
HGBL-MYC/BCL2瘤细胞弥漫性生长,形态多样,可呈BL样、母细胞样或介于DLBCL与BL之间的灰区形态。瘤细胞表达广谱B细胞抗原,绝大多数病例瘤细胞呈GCB免疫表型。诊断性分子异常需获得MYC和BCL2重排证据,可伴或不伴BCL6重排。但有MYC和BCL2重排的B细胞淋巴瘤不一定就是HGBL-MYC/BCL2。所有DLBCL-NOS在诊断前均推荐常规行MYC、BCL2和BCL6易位检测,至少需行MYC基因易位检测。
HGBL-11q瘤细胞形态与HGBL-MYC/BCL2有重叠,可呈BL样、母细胞样或介于DLBCL与BL之间的灰区形态。免疫表型:瘤细胞表达广谱B细胞标志物,CD10和BCL6阳性,Ki-67高表达(≥90%)。诊断性分子特征是MYC、BCL2、BCL6易位均阴性、11q23.3获得和11q24.1-端粒缺失,其中11q24.1-端粒缺失对诊断具有较高特异性,因此,当形态学、免疫表型符合HGBL-11q、又无MYC易位的病例,11q23.3获得阴性、11q24.1-端粒缺失阳性可诊断为HGBL-11q。
HIGBL-NOS代表一类异质性的侵袭性成熟B细胞淋巴瘤,瘤细胞弥漫性生长,可呈母细胞样、介于DLBCL与BL之间的灰区形态或BL样,常有“星空”现象。免疫表型:瘤细胞表达广谱B细胞标志物,大多数病例表达CD10、BCL6和BCL2,不恒定表达MUM1,大部分病例为GCB来源。HGBL-NOS必须排除HGBL-MYC/BCL2和HGBL-11q,形态学又不符合DLBCL-NOS的诊断要求,才能诊断为该类型。
原发纵隔大B细胞淋巴瘤
概述
原发纵隔大B细胞淋巴瘤(primary mediastinal Large B-cell Lymphoma,PMBCL)是DLBCL的特殊亚型之一,约占非霍奇金淋巴瘤的2%~4%,占DLBCL的6%~10%。PMBCL好发于年轻女性,男女之比为1:2,中位发病年龄为30~40岁。病变起源于胸腺髓质B细胞,常表现为前纵隔巨大肿块,可邻近侵犯到肺组织,常伴有上腔静脉综合征、胸腔或心包积液。大多数患者就诊时处于Ⅰ-Ⅱ期,初诊时大约80%患者的病变为局限累及,复发患者往往病变广泛。大部分PMBCL对化疗敏感,但强化免疫化疗方案疗效更好,可避免纵隔放疗引起的远期不良反应。
病理诊断
PMBCL的免疫表型与非特指型DLBCL相似,常表达B细胞相关抗原,如CD19、CD20、CD22、CD79a、PAX5和CD45,但常缺乏膜表面和胞质免疫球蛋白表达,更常见表达CD23、弱表达CD30,且多有PD-L1/2表达水平升高。
PMBCL的基因表达谱不同于非特指型DLBCL,而与经典型霍奇金巴瘤(CHL)有部分重叠。应注意与介于PMBCL和CHL之间的灰区淋巴瘤(GZL)进行鉴别。分子遗传学异常包括NF-KB、JAK/STAT通路异常活化,PD-L1/2扩增或9P24.1获得,以及MHC Ⅱ相关分子缺陷。最新研究发现,具有CD58突变的患者预后差,约占31%。高IPI评分伴有CD58突变者,5年PFS仅41%、5年OS为58%,特别是未采用强化方案治疗者预后更差,CD58野生型分别为76%和83%,而DUSP2突变患者预后相对较好。确诊PMBCL需要结合病理特征和临床表现进行整合判断。
原发中枢神经系统淋巴瘤
概述
原发中枢神经系统淋巴瘤(primary central nervous system lymphoma,PCNSL)是少见的非霍奇金淋巴瘤,好发于老年人,男性多于女性,中位发病年龄为65岁,95%以上患者病理类型为DLBCL。主要临床表现为颅内占位引起的头痛、感觉异常、运动障碍、神志异常等症状,小部分患者表现为脊髓及神经根病变。对仅累及视网膜、玻璃体等眼部结构的类型,称为原发眼内淋巴瘤,也属于PCNSL。立体定向导航脑组织穿刺活检是常用的诊断途径,部分患者通过术后获病理确诊。治疗以能透过血脑屏障的化疗药物为主,放疗为辅,手术不常规推荐。PCNSL预后差,5年生存率仅为29.9%。
病理诊断
立体定向活检是PCNSL常用的病理诊断方法,PCNSL大多为non GCB型,免疫组化呈PAX5、CD19、CD20、CD33和CD79a阳性,大多数细胞呈BCL6(60%~80%)和MUM1/IRF4(90%)阳性,而CD38和CD138呈阴性。在PCNSL中,CD10阳性率常低于10%,增殖指标Ki-67多在70%~90%之间,分子病理表型多数与活化B细胞淋巴瘤的MCD亚型相同。糖皮质激素会影响诊断,活检前应尽量避免或减少其使用。部分PCNSL患者出现脑脊膜或脑脊液播散,约15%患者脑脊液细胞学或流式细胞术检测阳性。
其他结外淋巴瘤
(一)原发乳腺弥漫大B细胞淋巴瘤
概述
原发乳腺弥漫大B细胞淋巴瘤(primary breast diffuse large B cell lymphoma,PB-DLBCL)是一种罕见的结外侵袭性NHL,占所有DLBCL的2.7%。临床表现为单侧乳房无痛性肿块,可伴有同侧引流区淋巴结增大。判断是否为原发乳腺淋巴瘤主要基于Wiseman和Liao提出的诊断四项标准,包括:部位位于乳腺,乳腺组织与淋巴瘤组织在解剖学位置上需要紧密相接;无乳腺淋巴瘤既往病史;诊断时不伴有广泛播散的淋巴瘤病灶;除区域淋巴结(同侧腋窝及锁骨上)受累外,无其他部位受累。治疗为以R-CIOP为主的整合治疗,PB-DLBCL有中枢受累的风险,有中枢症状者及时进行颅脑增强MR及脑脊液检查。
病理诊断
PB-DLBCL病理诊断时可行乳腺肿块切取/空芯针穿刺活检,也可行淋巴结完整切除或切取活检。组织形态及免疫表型与普通DLBCL NOS相同,因浸润乳腺小叶结构可呈假结节样生长,需要与浸润性小叶癌鉴别,特别是穿刺活检组织较少时。常规IHC包括CD20、CD3、CD5、CD10、BCL2、BCL6、MYC、Ki-67、IRF4/MUM1。流式细胞学检测包括κ/λ、CD3、CD5、CD19、CD10、CD20、CD45、TdT等。PB-DLBCL病理以non-GCB表型为主,约占60%~90%,MYD88(25%~70%)和CD79B(25%~40%)突变的发生率也很高。
(二)原发睾丸弥漫大B细胞淋巴瘤
概述
原发睾丸淋巴瘤(primary testicular lymphoma,PTL)是一种罕见淋巴瘤,DLBCI是PTL最常见的病理类型,占80%~98%。原发睾丸DLBCL(PT-DLBCL)占睾丸肿瘤的3%~9%,占NHL的1%~2%。中位发病年龄约66~68岁。少数患者合并HIV感染,好发于<50岁人群:大多数表现为单侧睾丸无痛性肿物或肿胀,少数表现为阴囊疼痛。诊断时双侧睾丸同时受累者约占6%~10%。尽管大多数患者的CNS-IPI评分较低,但对侧睾丸、中枢神经系统(CNS)是较常见的复发部位。
病理诊断
患侧睾丸切除术病理检查是确诊PTL的金标准。病理特点为异型淋巴细胞弥漫浸润于睾丸实质内,瘤细胞形似免疫母细胞或中心母细胞。免疫表型检测瘤细胞表达B细胞标志物CD19、CD20、PAX5等。EBER常为阴性。约60%~96%的PT-DLBCL患者为活化B细胞样(ABC)亚型。BCR通路相关基因具较高变率,如MYD88、CD79B突变等。部分患者出现9p24.1拷贝数改变和易位导致PD-L1/L2蛋白表达增加。
(三)原发骨淋巴瘤
概述
原发骨淋巴瘤(primary bone lymphoma,PBL)是指病变仅限于骨骼系统,或周围软组织浸润,但无全身症状的淋巴瘤。PBL是一种罕见且独特的结外淋巴瘤类型,占骨恶性肿瘤的5%~7%,恶性淋巴瘤的1%。病因尚未明确,研究报道可能与病毒感染、骨髓炎、创伤等相关。患者年龄分布较广,但多集中于45~60岁。PBL可累及全身骨骼,以四肢长骨、盆骨和脊柱最常见。相对于其他结外淋巴瘤预后尚可,5年生存率能达50%左右。由于该病早期无明显症状,常以疼痛和局部肿块就诊。累及脊柱的患者多以腰痛、腿痛甚至截瘫就诊,因此早发现且采取及时、正确治疗对患者生存预后和生活质量尤为关键。
病理诊断
DLBCL是PBL最常见的病理类型,约占所有亚型的80%。由于患者缺乏特异的影像学表现,易被误诊为骨肉瘤、尤文肉瘤和恶性肿瘤骨转移等,因此需要病理活检才能做出明确诊断。原发骨DLBCL(PB-DLBCL)常表达成熟B细胞的免疫标记,包括CD20、CD19、PAX5和CD79a等,BCL2和BCL6的表达阳性率高。T细胞标志物常阴性。非GCB亚型比GCB亚型更为常见。PB-DLBCL存在多种遗传学异常,BCL2、BCL6和MYC重排的发生率分别为19%、14%和9%,但BCL2及c-MYC同时易位很少见。
(四)原发皮肤淋巴瘤
概述
原发皮肤淋巴瘤(primary cutaneous lymphoma,PCL)是一组罕见的淋巴瘤,包括原发皮肤B细胞淋巴瘤(primary cutaneous B cell lymphoma,PCBCL)、蕈样霉菌病/Sezary综合征(Mycosis Fungoides/Sezary Syndrome,MF/SS)及原发皮肤CD30+T细胞淋巴增殖性疾病(primary cutaneous CD30+T-cell lymphoproliferative disorders).
病理诊断
(1)原发皮肤B细胞淋巴瘤
PCBCL包括三种亚型:原发皮肤滤泡中心淋巴瘤(primary cutaneous follicle center lymphoma,PCFCL)、原发皮肤边缘区淋巴瘤(primary cutaneous marginal zone lymphoma,PCMZL)、原发皮肤弥漫性大B细胞淋巴瘤-腿型(primary cutaneous diffusclarge B cell lymphoma,PCDLBCL,leg type)。PCBCL的病理诊断需对皮损部位进行穿刺/切取/切除活检,不建议进行刮取活检。
PCFCL是最常见的PCBCL亚型。可呈滤泡、滤泡/弥漫混合或弥漫性生长,以中心细胞为主,混杂多少不等的中心母/免疫母细胞。免疫表型:表达B系标记如CD20、CD79a和BCL6;表面Ig阴性。弥漫性生长类型者CD10为阴性。BCL2为阴性,或呈弱表达。若CD10和BCL2强表达,或BCL2重排时,需排除经典型(淋巴结性)滤泡性淋巴瘤(FL)皮肤受累。弥漫性生长的PCFCL常表现为形态单一的大中心细胞样细胞,可混有多少不等的中心母细胞,且Ki-67指数较高,不要误认为是DLBCL。
PCMZL为次常见亚型,具边缘区淋巴瘤病理形态学特征。免疫表型:CD10和BCL6阴性,BCL2为阳性。约1/3病例可表达IgG4。根据Ig重链重排可分为两组,预后不同:①CXCR3阴性和Ig类别转换亚型(IgG、IgA和IgE),以大量反应性T细胞及结节外周多量簇状单形性浆细胞为特征;②CXCR3阳性和IgM阳性(非类别转换)亚型较少见,浆细胞呈少量散在分布,反应性T细胞少,可能有皮肤外侵犯。Ig类别转换亚型是一种克隆性慢性淋巴增殖性疾病,病程缓慢。
PCDLBCL-腿型为最罕见亚型,由中心母细胞和免疫母细胞样细胞组成,无论发生在人体皮肤的任何部位,都称为PCDLBCL-腿型:免疫表型:表达B表型如CD20、CD79a等,以及单克隆免疫球蛋白、BCL2(强)、MUM1/IRF4、FOXP1、MYC:而CD10阴性。基因表达谱:PCDLBCL通常为活化B细胞(ABC)亚型。应注意与弥漫性PCFCL鉴别,后者表现为大的中心细胞样细胞,可混杂多少不等的中心母细胞样细胞,表达BCL6,不表达MUM1:FISH检测:MYC、BCL6基因易位常见。
用于诊断及鉴别诊断的免疫组化应包括:CD20,CD3,CD10,BCL2,BCL6,IRF4/MUM1;此外,为了明确亚型,还应包括:Ki-67,CD5,CD43,CD21,CD23,cyclin D1,kappa/lambda。为鉴别DLBCL(腿型)及原发皮肤滤泡淋巴瘤,需评估IgM、IgD、IgA、IgG、IgE及FOXP1。建议完善EBER检查。若怀疑为系统性FL,需进行t(14:18)检测。
(2)MF/Sezary综合征
MF是最常见的皮肤T细胞淋巴瘤(CTCL),有许多临床病理变异型。即使在具有典型特征的病例中,MF的组织病理学表现也需与临床表现相关联,才能做出明确诊断。斑片性病变通常很难确诊,因此,需行多次皮肤活检。进行皮肤活检之前,建议局部治疗至少停2~3周。色素沉着或色素脱失等特殊的临床病理变异有助于诊断。斑块期和肿瘤期病变需注意与其他类型淋巴瘤鉴别,结合临床表现及病史非常关键。
MF的瘤细胞CD3+、CD4+、CD45RO+、CD8-,即所谓的皮肤驻留记忆T细胞。在少数MF病例中,可能会出现CD4-、CD8+成熟T细胞表型或更罕见的γ/δ T细胞表型(BF1-、TCR γ/δ+、CD3+、CD4-、CD8+)。此类病例具有与CD4+者有相同的临床表现和预后,不应孤立考虑。异常表型(如CD2、CD3和CD5等泛T细胞抗原的缺失)是MF诊断的重要辅助证据,但在MF早期少见。
大细胞转化(LCT)在组织学上定义为转化大细胞占比超过25%。
Sezary综合征为CTCL的白血病变异型,与MF关系密切,但具有独有的特征。SS很少见,占皮肤淋巴瘤5%以下,多见于老年人。SS的特征是皮肤中存在Sezany细胞。异常T细胞(>1000个异常细胞/μL)通过细胞病理学或流式细胞术定义为Sezary细胞(异常亚群包括但不限于CD4+CD7-或CD4+CD26-细胞:TRBC1有助于检测克隆性,尤其是在CD7或CD26未丢失的情况下)。SS来自胸腺记忆T细胞,而皮肤驻留效应记忆T细胞是MF的起源细胞。这表明SS发病与MF不同。临床上可见这两种表现重叠的情况。
MF有许多变异型,主要包括亲毛囊性MF(Folliculotropic mycosis fungoides)、派杰样网状细胞增生症(Pagetoid reticulosis)和肉芽肿性皮肤松弛症(Granulomatous slack skin)等。
(3)原发皮肤CD30+T细胞淋巴增殖性疾病
原发性皮肤CD30+T细胞淋巴增生性疾病(LPD)包括原发皮肤间变性大细胞淋巴瘤(ALCL)、淋巴瘤样丘疹病(LyP)以及临床和组织病理学特征重叠的“交界性”病例。
临床特征与组织病理学特征的相关性对诊断LPD至关重要;仅凭病理检查难以做出精确诊断。建议进行完整的皮肤检查以除外MF。为确定诊断,至少需要包括以下的免疫组化:CD3、CD4、CD8、CD20、CD30、CD56、ALK。其他免疫组化包括CD2、CD5、CD7、CD25、TIA1、颗粒酶B、穿孔素、IRF4/MUM1、EMA、TCRβ、TCRδ。EBER原位杂交。用于检测TCR基因克隆性重排或其他克隆性评估的分子分析。原位杂交:alk和dusp22基因重排。建议对血清HTLV-1/2进行检测,其结果可能影响治疗方案。
滤泡性淋巴瘤
概述
滤泡性淋巴瘤(follicular lymphoma,FL)是一类起源于滤泡中心B细胞的非霍奇金淋巴瘤(NHL),典型免疫表型为CD5-CD10+CD19+,伴t(14:18)(q32;q21),临床呈高度异质性。我国FL的发病率占B细胞NHL的8%~23%,低于欧美地区。FL发病率从35岁开始逐步增加,至70岁达峰值。国内资料显示FL诊断时中位年龄约53岁,女性发病率略高于男性,5年PFS率及OS率分别为61%和89%。
病理诊断
FL的病理诊断标准主要根据形态学、免疫组化检测来诊断,必要时进行流式细胞学、分子遗传学检测辅助诊断。形态学上多数FL有明显的滤泡结构,这些肿瘤性滤泡部分融合,缺乏外套层,失去极向和星空现象。肿瘤性滤泡由中心细胞和中心母细胞组成,前者细胞小到中等大小,核细长、扭曲或有裂沟,核仁不明显,胞浆少而空亮,后者一般为中等或大细胞,核圆形或卵圆形,也可不规则,染色质空泡状,有1~3个靠近核膜的核仁,胞浆少。FL可根据中心母细胞数量的多少进一步分级。
滤泡性淋巴瘤的分级
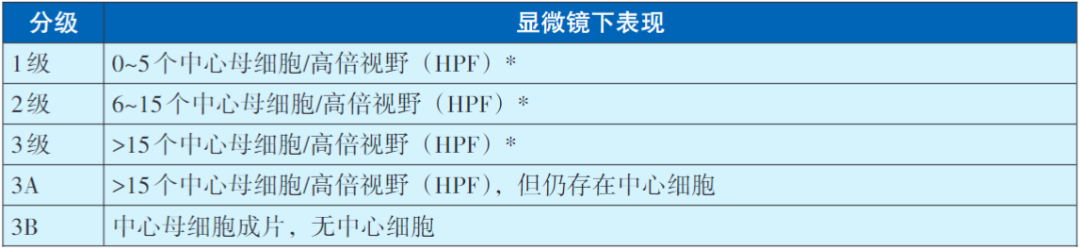
瘤细胞常表达表面免疫球蛋白(sIg)和B细胞相关抗原(CD19,CD20,CD22,CD79a,PAX5),此外,尚表达生发中心相关标记,如CD10,BCL6,GCET1,HGAL(GCET2),LMO2等。CD10和BCL6在滤泡中的表达往往比滤泡间区瘤细胞更强。CD10及BCL6阴性的FL诊断需有表达其他生发中心标记的证据支持。大多数FL病例(约85%)表达BCL2,BCL2阳性有助于区别滤泡性淋巴瘤与滤泡反应性增生,但在鉴别FL,与其他低度恶性的B细胞淋巴瘤中无价值。滤泡中存在CD21(+)和CD23(+)的FDC网。基因重排检测可见Ig重链和轻链基因的克隆性重排,可变区基因存在广泛的体细胞突变并有克隆间的异质化,提示来源于生发中心细胞。
几乎所有的滤泡性淋巴瘤均有细胞遗传学异常。最常见的为t(14;18)(q32;q21),即IGH/BCL2基因融合,发生率为70%~95%,可以用FISH方法检测。
套细胞淋巴瘤
概述
套细胞淋巴瘤(mantle cell lymphoma,MCL)是一种少见的B细胞起源非霍奇金淋巴瘤。西方国家中MCL约占成人NHL的3%~10%,在中国MCL约占B细胞淋巴瘤的6.3%。MCL兼具惰性和侵袭性淋巴瘤的特点,侵袭性较强,临床分期较晚,结外浸润广泛,对传统放化疗不敏感,预后较差。男女比为2~3:1,西方国家诊断的中位年龄约68岁,国内约60岁。目前仍无法治愈,未观察到生存曲线平台,几乎所有患者出现复发。
病理诊断
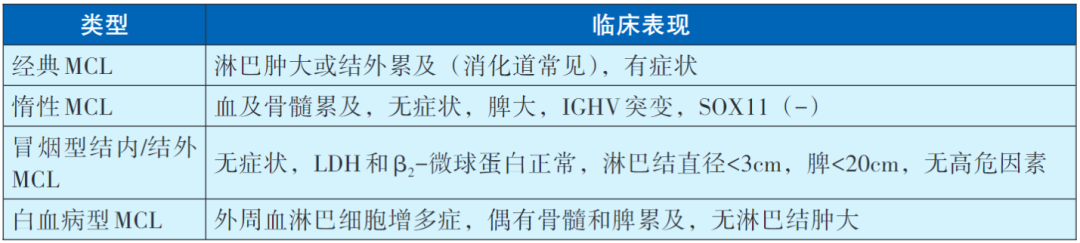
MCL的诊断主要基于组织病理学检查,包括经典型、多形性和母细胞变异型,后两种类型为侵袭性亚型。形态上MCL生长模式可以是结节状、弥漫性,也可以是套区生长模式。典型免疫学表型特征为CD19(+),CD20(+),CD22(+),CD43(+),CD79a(+),CD5(+),FMC7(+),slgM/slgD(+++),CD23(-),CD10(-),CD200(-)和BCL6(-)。病理特征为t(11;14)(q13;q32)和cyelin D1过表达。约5%的MCL cyclinD1(-),需要FISH进一步证实,如仍然阴性,则需要加做CCND2和CCND3(2B)。WHO-HAEM5中MCL分类,主要有两种类型,一种是由成熟B细胞组成非生发中心的经典性MCL,该型无或有极少IGHV突变,有转录因子SOX11突变,临床表现为淋巴结和结外部位累及,侵袭性较强:另一型是比较少见(10%~20%)的非淋巴结性白血病型MCL(leukemic non-nodal MCL),该型是一种起源于生发中心的惰性淋巴瘤,IGHV会发生高频突变,且转录因子SOX11不表达或极少表达,临床表现为外周血、骨髓和脾脏受累。接受传统治疗的MCL中,TP53突变提示预后更差,故行TP53基因检测有助判断预后。
MCL分型及临床表现
边缘区细胞淋巴瘤
概述
边缘区淋巴瘤(marginal zone lymphoma,MZL)是一组异质性较强的惰性淋巴瘤,包括黏膜相关淋巴组织(mucosa-associated lymphoid tissues,MALT)淋巴瘤也称为结外边缘区淋巴瘤(extranodal marginal zone lymphoma,EMZL)、结内边缘区淋巴瘤(nodal marginal zone lymphoma,NMZL)及脾边缘区淋巴瘤(splenic marginal zonelymmphoma,SMZL)三种亚型。三者在形态学、免疫表型和基因表型方面基本相似但其临床表现和治疗选择略有差异。胃肠道是MALT淋巴瘤最常见的原发部位,约占所有MALT淋巴瘤的50%,其他常见部位包括眼附属器、腮腺、肺部、甲状腺和皮肤等,约15%~20%的患者存在骨髓受侵。大部分MALT淋巴瘤为局限性疾病,约1/3的患者表现为播散性。MZL的病因与慢性感染或炎症所致的持续免疫刺激密切相关。胃MALT淋巴瘤与幽门螺杆菌(Hp)的慢性感染有关,小肠MALT淋巴瘤与空肠弯曲菌感染有关,22%~35%的淋巴结MZL、脾脏MZL和非胃MALT淋巴瘤中存在丙型肝炎病毒(HCV)感染。其他感染还包括与结膜和眼附属器MZL相关的鹦鹉热衣原体、与皮肤结外MZL相关的伯氏疏螺旋体和支气管结外MZL相关的木糖氧化无色杆菌。同时合并自身免疫性疾病及应用免疫抑制剂者也会增加MZL的发病率。甲状MALT淋巴瘤与桥本氏甲状腺炎有关,腮腺MALT淋巴瘤与干燥综合征有关,约15%的原发性肺MALT淋巴瘤合并有自身免疫病,包括多发性硬化、系统性红斑狼疮等,特别是干燥综合征,都是肺MALT淋巴瘤的危险因素。
病理诊断
MZL的病理诊断标准主要根据形态学和免疫组化的方法来诊断,必要时进行流式细胞的检测。形态学特征包括淋巴结和脾脏的生发中心缩小、边缘区增宽。MZI典型的免疫表型为CD5(-)、CD10(-)、CD20(+)、CD21(±)、CD23(±)、CD43(±)、Cyclin D1(-)以及BCL2(-)。t(11;18)、t(1;14)、t(14;18)和t(3;14)是MALT中比较常见的染色体改变。对于SMZL,也可检测-7q+、3q等染色体异常或NOTCH2、KLF2等基因突变,此外,还可通过检测MYD88突变和淋巴浆细胞淋巴瘤/华氏巨球蛋白血症(LPL/WM)鉴别,以及检测BRAF突变与毛细胞白血病进行鉴别。
慢性淋巴瘤细胞白血病/小淋巴细胞淋巴瘤
概述
慢性淋巴细胞白血病(CLL)/小淋巴细胞淋巴瘤(SLL)是一种成熟B淋巴细胞克隆增殖性肿瘤,临床表现外周血淋巴细胞增多、肝脾及淋巴结肿大,并累及淋巴系统以外其他器官,晚期可表现为骨髓衰竭。CIL与SLL具有同样的病理和免疫表型特点。不同的是,CIL疾病主要集中在外周血中,而SLL疾病主要集中在淋巴结。CLL/SIL是西方最多见的白血病类型,占到全部白血病的25%~35%,欧美人群中年发病率达到4~5/10万。男性多见,男女比例1.2~1.7:1。而亚洲人群CLL/SLL的发病率明显低于欧美。日本、韩国等地人口登记资料显示的发病率大约是欧美的十分之一。CLL/SIL老年发病,欧美报告的中位发病年龄在70~75岁,而我国的中位发病年龄为60~65岁。
诊断
慢性淋巴细胞白血病的诊断需要满足以下诊断标准:
达到以下3项标准可以诊断CLL:①外周血单克隆B淋巴细胞计数≥5x109/L,且持续≥3个月(如具有典型的CLL免疫表型、形态学等特征,时间长短对CLL的诊断意义不大);②外周血涂片特征性的表现为小的、形态成熟的淋巴细胞显著增多,其细胞质少、核致密、核仁不明显、染色质部分聚集,并易见涂抹细胞;外周血淋巴细胞中不典型淋巴细胞及幼稚淋巴细胞≦55%;③外周血典型的流式细胞学免疫表型:CD19+、CD5+、CD23+、CD200+、CD10-、FMC7-、CD43+/-;表面免疫球蛋白(sIg)CD20、CD22及CD79b的表达水平低于正常B细胞(dim)。流式细胞学确认B细胞的克隆性,即B细胞表面限制性表达κ或λ轻链(κ:λ>3:1或<0.3:1)或>25%的B细胞sIg不表。
SLL与CLL是同一种疾病的不同临床表现,约20%的SLL进展为CLL。淋巴组织具有CLL的细胞形态与免疫表型特征,确诊必须依赖病理组织学及免疫组化检查。临床特征:①淋巴结和(或)脾、肝肿大;②无血细胞减少:③外周血单克隆B淋巴细胞<5x109/L。CLL与SLL的主要区别在于前者主要累及外周血和骨髓,而后者则主要累及淋巴结和骨髓(此特征很重要,对骨髓受累SLL患者可以利用骨髓标本进行流式细胞术免疫分型、染色体核型分析、基因突变等检测)。Lugano Ⅰ期SLL可局部放疗,其他SLL的治疗指征和治疗选择同CLL
单克隆B淋巴细胞增多症(MBL):是指健康个体外周血存在低水平的单克隆B淋巴细胞。诊断标准:①B细胞克隆性异常;②外周血单克隆B淋巴细胞<5x109/L;③无肝、脾、淋巴结肿大(淋巴结长径<1.5cm);④无贫血及血小板减少;⑤无慢性淋巴增殖性疾病(CLPD)的其他临床症状。根据免疫表型分为3型:CLL样表型、不典型CLL样表型和非CLL样表型。对于后二者需全面检查,如影像学、骨髓活检等,以排除外周血受累的非霍奇金淋巴瘤。对于CLL样表型MBL,需根据外周血克隆性B淋巴细胞计数分为“低计数”MBL(克隆性B淋巴细胞<0.5x109/L)和“高计数”MBL(克隆性B淋巴细胞≥0.5x109/L),“低计数”MBL无需常规临床随访,而“高计数”MBL的免疫表型、遗传学与分子生物学特征与Rai 0期CLL接近,需定期随访。几乎所有的CLL来自CLL表型的MBL,所以确诊的CLL患者,应尽可能追溯既往血细胞变化,可以初步了解疾病进展速度。对于非CLL表型的MBL,应进行包括影像学在内的系统检查,以排除其他外周血受累的非霍奇金淋巴瘤。
鉴别诊断
根据外周血淋巴细胞计数明显升高、典型的淋巴细胞形态及免疫表型特征,大多数CLL容易诊断,但尚需与其他疾病,特别是其他B慢性淋巴细胞增殖性疾病(B-CLPD)相鉴别。根据CLL免疫表型积分系统(CD5+、CD23+、FMC7-、sIgdim、CD22/CD796dim/-各积1分),CLL积分为4~5,其他B-CLPD为0~2分。积分≦3分的患者需要结合淋巴结、脾脏、骨髓组织细胞学及遗传学、分子生物学检查等进行鉴别诊断,特别是套细胞淋巴瘤(MCL)、白血病期的边缘区淋巴瘤(MZL)(尤其是脾边缘区淋巴瘤(SMZL))、淋巴浆细胞淋巴瘤(LPL),一般不同时表达CD5和CD23。
大多数CLL,细胞表达CD5(表达强度低于T细胞,临床上需注意假阴性可能)和B细胞抗原CD19、CD20和CD23。典型的CLL免疫表型为CD5+、CD23+、CD200+、CD43+/-、CD10-、CD19+、CD20dim(dim:弱表达)、sIgdim和Cyclin D1-(此抗原需通过免疫组织化学检测);部分患者可能表现为sIgbright(bright:强表达)、CD23-/dim、FMC7弱阳性。由于同样是CD5+的MCL,FMC7+、CD23-,sIg及CD20表达强于CLL,与CLL有类似的免疫表型,因此对于免疫表型不典型的CLL(CD23dim或阴性、CD20bright、sIgbright或FMC-7+等),需要采用免疫组织化学染色检测Cyclin D1、SOX11、LEF1等(CLL表达LEF1,MCL表达Cyelin D1及SOX11)以及FISH检测t(11;14),以便与MCL鉴别。CD200+可用于区分CLL和MCL,后者通常为CD200-。
外周T细胞淋巴瘤
概述
外周T细胞淋巴瘤(peripheral T-cell lymphoma,PTCL)是一组起源于成熟T细胞的侵袭性肿瘤性疾病,在中国约占NHL的20%以上,显著高于欧美的5%~10%。根据WHO 2022年血液肿瘤分类,T细胞淋巴瘤和NK细胞淋巴瘤归统在一个大类,根据主要累及部位,成熟T细胞淋巴瘤可表现为骨髓和外周血侵犯为主(白血病样)、原发皮肤、原发胃肠道、原发肝脾及淋巴结受累为主。除非特别指出,一般讲的PTCL,主要指以淋巴结受累为主的、包括淋巴结TFH细胞淋巴瘤(血管免疫母细胞型、滤泡型、非特指型)、间变性大细胞淋巴瘤(ALK阳性、AIK阴性或乳腺植入物相关性)和PTCL非特指型(PTCL-NOS)。原发肠道和原发肝脾的T细胞淋巴瘤也常参照一般PTCL治疗但效果不佳,而原发皮肤和白血病样表现的T细胞淋巴瘤,其诊治原则差别较大,在此不做介绍。
病理诊断
PTCL的诊断主要依赖病理形态结合免疫组化特征,有时需参考分子细胞遗传学检测和临床表现。病理形态是淋巴瘤诊断的基础,一般表现为肿瘤细胞弥漫浸润,破坏淋巴结或受累组织的正常结构,瘤细胞可大小不等,异型性明显。免疫组化染色是PTCL诊断不可或缺的检测手段。一般诊断所需免疫组化标志物有CD2、CD3、CD4、CD5、CD7、CD8、CD10、CD20、CD30、CD43、CD56、PD1、CXCL13、ALK、TIA-1、granzyme B、Ki-67、PAX5或CD19、CD21等。考虑nTFHL时可加做ICOS和BCL6等。PTCL的病理诊断中宜常规进行EBER-ISH检测。当肿瘤与反应性增生难以鉴别时,可参考TRβ、TRγ基因重排检测结果,但重排阳性也可出现在部分反应性增生病例中。不同类型的PTCL有其各自的病理特征和免疫特点。
血管免疫母细胞淋巴瘤(AITL;WHO第五版称为淋巴结TFH细胞淋巴瘤,血管免疫母细胞型;简称nTFHL-AI):①淋巴结内多形性细胞浸润,伴有明显的高内皮小静脉和滤泡树突细胞增生。病变组织中的细胞成分复杂多样,肿瘤性T细胞背景下,常伴有多克隆甚至单克隆大B细胞增生,部分病例在复发时表现为弥漫大B细胞淋巴瘤。早期患者易误诊为反应性增生。②瘤细胞除表达CD3和CD4等T细胞标志外,应还表达至少两种或以上TFH标志,如PD1,ICOS,CXCL13,CD10和BCL6。③分子遗传学上,TET2、DNMT3A、RHOA和IDH2突变发生率较高,第五版WHO分类将RHOA和IDH-突变写入了nTFHL诊断的理想条件中。
间变大细胞淋巴瘤(ALCL):瘤细胞大,呈多形性,大多数细胞表达CD30,常不同程度地缺失T细胞标记,如CD3等。ALK+ALCL多数由t(2;5)(p23;q35)易位,导致NPM1/ALK融合,而致ALK蛋白过表达。典型病例的诊断常无需进行融合基因检测。ALK-ALCI预后较差,TP63重排和TP53缺失者预后差,DUSP22重排的预后意义仍有争议。
PTCL-NOS:对不能满足已单列的各种类型PTCL的病例则归入PTCL-NOS。该类肿瘤以淋巴结受累为主,但仍有较大异质性。
其他少见类型:如肠道T细胞淋巴瘤,我国以单形性嗜上皮性肠道T细胞淋巴瘤(MEITL)常见,免疫组化多数表现为CD3+,CD5-,CD4-,CD8+,CD30-,CD43+、CD56+,CD57-,TIA-1+,EBER-。肠病相关T细胞淋巴瘤(EATL)常表现为CD8-,CD56-,CD30+
鉴别诊断:
①PTCL不同亚型之间的鉴别;②NK/T细胞淋巴瘤;③反应性淋巴增生。
结外NK/T细胞淋巴瘤
概述
结外NK/T细胞淋巴瘤(extranodal NK/T cell lymphoma,ENKTL)是侵袭 NHL的一种独特亚型,在一些亚洲、拉丁美洲国家中患病率高。主要发生在上呼吸消化道,包括鼻腔、鼻咽、鼻窦、扁桃体、下咽和喉部,临床上将其称为鼻型NK/T细胞淋巴瘤;约10%~20%的淋巴瘤发生在非鼻腔部位,如皮肤、睾丸、胃肠道等,称为非鼻型NK/T细胞淋巴瘤。结外NK/T细胞淋巴瘤的常见症状包括鼻塞、鼻出血、发热、面部浮肿和颈部肿块,鼻外NKT细胞淋巴瘤临床侵袭性更强。ENKTL发病年龄常为40~50岁,以男性为主,男女比例为2~3:1。分期方面,约70%~90%的患者为Ⅰ期或Ⅱ期淋巴瘤。
病理诊断
ENKTL,的病理学特征为瘤细胞弥漫性浸润,呈血管中心性/血管破坏性生长,致受累组织缺血坏死及黏膜溃疡。ENKTL诊断所需免疫组化标志物包括CD3、CD56、CD2、CD4、CD5、CD7、CD8、CD20、PAX5、TIA-1、granzyme B、Ki-67。必做EBER-ISH。ENKTL的典型免疫表型为CD2(+)、cCD3ε+(surface CD3-)、CD5(-/+)、CD56(+),细胞毒性分子如TIA-1、granzyme B及perforin阳性,EBER-ISH(+)。EBER-ISH阴性时诊断宜谨慎,如CD56(+)、CD3(+)、细胞毒标志物均表达时,可诊断为ENKTL。60%~90%的ENKTL缺乏TR基因重排。
鉴别诊断
①其他成熟T/NK细胞来源肿瘤,如PTCL-NOS、ALCL,以及少见的侵袭性NK细胞白血病等;②发生于儿童青少年的病例应与儿童系统性EBV阳性T细胞淋巴瘤相鉴别:③少数病例还需注意与EBV+的癌相鉴别,应增加CK和EMA等上皮标志物检测.
近期有研究根据分子生物学特征将ENKTL分为TSIM、MB和EHA三种亚型,根据肿瘤免疫微环境将ENKTL分为免疫耐受、免疫逃逸-A、免疫逃逸-B和免疫沉默四种亚型,这可能为靶向、免疫治疗提供分子基础。
伯基特淋巴瘤
概述
伯基特淋巴瘤(burkitt lymphoma,BL)是一种少见、高度侵袭性的非霍奇金淋巴瘤,常累及结外部位。根据发病特征分类,我国BL发生具有“散发性”和“免疫缺陷相关性”的特点,部分与EBV、HIV感染和异基因移植相关的免疫缺陷有关。BL还有特征性遗传学异常,包括位于8号染色体的MYC基因与位于14号染色体的免疫球蛋白重链可变区(IGHV)重排,即t(8;14),或与位于2号、22号染色体上的免疫球蛋白轻链基因重排,即t(2;8)、t(8;22)。在治疗方面,BL患者需要接受预后分层指导下的增强剂量化疗。目前,规范诊疗可使约60%的患者获得持续缓解。
病理诊断
经典型BL形态学表现为较均一的中等大小肿瘤性B细胞弥漫增生,核圆形,有小核仁,核分裂象及凋亡易见,细胞质中等量,常含有脂质空泡。增殖指数近100%,标本中吞噬凋亡细胞核碎片的大量反应性巨细胞可能形成“星空”现象。瘤细胞呈成熟生发中心B细胞的免疫表型:CD19、CD20、CD79a、PAX5、CD10、BCL6和MYC呈阳性;CD5、BCL2和TdT常呈阴性。MYC易位普遍存在,需注意MYC易位也可见于其他类型肿瘤(例如:高级别B细胞淋巴瘤),伯基特淋巴瘤多无BCL2易位。多数散发性BL EBV阴性,地方性BL及免疫缺陷相关性BL常EBV阳性。WHO-HAEM第5版修订建议对EBV阳性BL和EBV阴性BL这两种亚型进行区分,与EBV阴性BL相比,EBV阳性BL的体细胞超突变水平更高。
2016年 WHO造血与淋巴系统肿瘤分类新提出“伴11q异常的伯基特样淋巴瘤”,其基因表达谱和形态与经典BL类似,但无MYC基因异常,而是具有11q染色体改变。因基因突变特点更接近于大B细胞性淋巴瘤,WHO-HAEM第5版修订将该病更名为“伴11q异常的高级别B细胞淋巴瘤”。
霍奇金细胞淋巴瘤
概述
霍奇金淋巴瘤(Hodgkin lymphoma,HL)是一种累及淋巴结和淋巴系统的恶性肿瘤。我国HL的发病率明显低于欧美国家,占全部淋巴瘤的8.54%,男性多于女性。我国HL发病年龄较小,年龄-发病曲线呈现单峰,高峰在40岁左右。90%的HL以淋巴结肿大为首发症状,以颈部淋巴结和锁骨上淋巴结常见,然后扩散至其他淋巴结晚期可侵犯血管,累及脾、肝、骨髓和消化道等。WHO将HL分为2个主要类型,包括经典霍奇金淋巴瘤(CHL)和结节性淋巴细胞为主的霍奇金淋巴瘤(NLPHL)CHL约占HL的90%,CHL的特征是在炎症背景下存在Reed-Stemnberg细胞,而NLPHI,缺乏Reed-Sternberg细胞,其特征是存在淋巴细胞为主的细胞,有时称为爆米花细胞。其中CHL又可分为四个亚型,即结节硬化型,混合细胞型,淋巴细胞耗竭型以及富含淋巴细胞型。我国HL以混合细胞型居多
HL的病因和发病机制尚不明确,可能与遗传背景、EB病毒感染、免疫抑制、电离辐射及基因突变等相关。在过去几十年中,HL的治疗取得了显著进展;对大部分患者,已成为可治愈的恶性肿瘤。需综合疾病特点、一般情况、经济、社会和治疗药物等综合因素考虑个体化、多学科整合诊治(MDT to HIM),是进一步提高疗效和长期生存质量的关键。
病理诊断
经典霍奇金淋巴瘤(CHL)
CHL根据背景的细胞成分、HRS细胞形态不同及组织构象特征可分为4个亚型:结节硬化型(NS)、混合细胞型(MC)、淋巴细胞丰富型(LR)、淋巴细胞消减型(LD)。这些亚型在发病部位、临床特征、生长方式、纤维化、背景反应性细胞的组成、瘤细胞数量和非典型程度及EBV的感染率有所不同,但瘤细胞的免疫表型是相同的。CHL免疫表型包括CD15(+/-)、CD30(+)、PAX-5弱阳性(少数病例可阴性或强阳性),以及CD3(-)、CD20(-)(或弱阳性、异质性阳性)、CD45(-)、CD79a(-)、BOB.1和0ct-2 至少一个失表达。
结节性淋巴细胞为主型霍奇金淋巴瘤(NLPHL)
常以小淋巴细胞结节状或结节状弥漫性增殖为特征,伴有单个散在的大瘤细胞,称为淋巴细胞为主型(LP)或爆米花细胞。LP细胞被PD1/CD279+T细胞包围。NLPHL免疫表型包括CD20(+)、CD45(+)、CD79a(+)、BCL6(+)、PAX-5(+),BOB.1和0ct-2均阳性,以及CD3(-)、CD15(-)、CD30(-)。诊断时需完善CD3、CD15、CD20、CD21、CD30、CD45以及CD57。形态学和免疫组化是诊断HL的关键方法,对诊断不明者可能需要更多的分子标记物检测。
Castleman病
概述
Casteman病(Castleman disease,CD)曾被称为巨大淋巴结病或血管滤泡性淋巴结增生症,是一组罕见、具有特征性组织病理学特点的异质性淋巴组织增生性疾病,被纳入中国第一批罕见病目录。目前认为白细胞介素6(intereukin-6,IL-6)可能是其疾病驱动因素,部分病例与人类疱疹病毒-8(human herpesvirus-8,HHV-8)感染关系密切。病理形态上,CD可分为透明血管型(hvaline vascular,HV-CD)、浆细胞型(plasmacytic,PC-CD)及混合型(Mixed-CD)。HV-CD以生发中心萎缩和过度血管化为病理特征,PC-CD的特征是生发中心增生和多型浆细胞增多,Mixed-CD兼具上述两种特征。
由于CD的罕见性,临床及病理诊断相对困难,加之对该病发病和转归尚不清楚,需要有规范化的诊断和治疗,以提高CD患者整体诊断率及疗效。
病理诊断
HV-CD:淋巴结被膜增厚、玻璃样变,淋巴窦消失。淋巴滤泡增生,滤泡套区增宽,滤泡生发中心萎缩,可见一套区包绕多个生发中心的结构(进行性转化的生发中心,PDGC)。萎缩的生发中心主要由梭形滤泡树突细胞和血管内皮细胞组成,生发中心内的血管内皮细胞增生、玻璃样变。套区小淋巴细胞围绕着萎缩的生发中心呈“葱皮”样排列。滤泡间区内见小血管增生,管壁透明变。可见小血管垂直插入萎缩的生发中心,呈“棒棒糖”样改变。淋巴结周围组织血管旁常见纤维化和硬化改变。
PC-CD:可见HV-CD样淋巴滤泡,但部分病例或部分病灶的滤泡生发中心萎缩不明显,甚至会出现生发中心增生和扩大,可见嗜伊红物质沉积。滤泡间区和髓索内见明显增多的多克隆性浆细胞,常见Russel小体,也可见散在的嗜酸性粒细胞和肥大细胞浸润。与HV型相似,其滤泡间区可见血管透明变,但该特点没有HV型明显。有时可见滤泡套区的“葱皮”样改变。
Mixed-CD:形态特点兼具HV-CD及PC-CD的特征
Castleman病的病理诊断
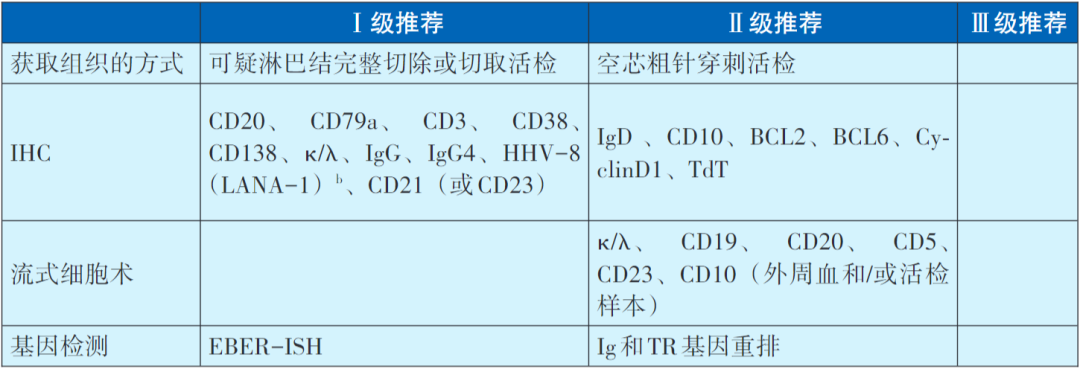
注:a.CD的诊断主要基于组织病理学检查,含形态学和免疫组化染色,必要时参考流式细胞术及基因检测结果。优选完整淋巴结切除活检,若无法进行,可行粗针穿刺活检明确病理诊断。b.可根据淋巴结组织病理的LANA-1免疫组化染色和(或)外周血中HHV-8 DNA检测结果判断是否为HHV-8阳性,如果前述两项检测中任一项阳性,可诊断为HHV-8阳性MCD;若无HHV-8感染证据,则诊断为HHV-8阴性MCD。
参考文献及书籍:
1.《2025 中国肿瘤整合诊治指南(CACA)——淋巴瘤》
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言











#淋巴瘤# #CACA#
4