

通过微创液体活检分析循环肿瘤DNA (ctDNA),有望早期发现多癌和监测微小残留疾病。大多数液体活检的检测技术采用靶向ctDNA深度测序,并将检测限制在最常见的癌症类型和SNV。一些研究将这些信息与靶向表观遗传分析和蛋白质标记结合起来,以提高对更大范围肿瘤类型的敏感性。组织特异性甲基化模式可用于检测ctDNA中的癌症信号和起源组织,然而,亚硫酸处理可破坏高达80%的可用ctDNA,使SNV无法调用,这些限制导致现有的方法很难整合多种数据模式。
近日,杂志Nature communications上发表了一篇题为“Multimodal cell-free DNA whole-genome TAPS is sensitive and reveals specific cancer signals”的文章。作者开发了一种使用深度全基因组TAPS的ctDNA检测方法,可在单次测序中同时分析基因组和甲基化组学数据。作者对有症状患者的多种癌症类型进行诊断准确性研究,结果显示达到了94.9%的敏感性和88.8%的特异性。即使在ctDNA分数低至0.7%时,也具有很强的辨别能力(86% AUC)。

图片来源:Nature communications
主要内容
TAPS原理概述
TET辅助吡啶硼烷测序(TAPS)(TET-Assisted Pyridine Borane Sequencing)是一种具有单碱基分辨率的测序方法,用于检测5-甲基胞嘧啶和5-羟基甲基胞嘧啶。与亚硫酸测序不同,TAPS是一种破坏性较小的方法,它采用TET酶与硼烷的组合仅转化5%的甲基化胞嘧啶,从而保留基因组信息,并为在相同测序数据上同时进行甲基组和基因组分析提供了可能性。
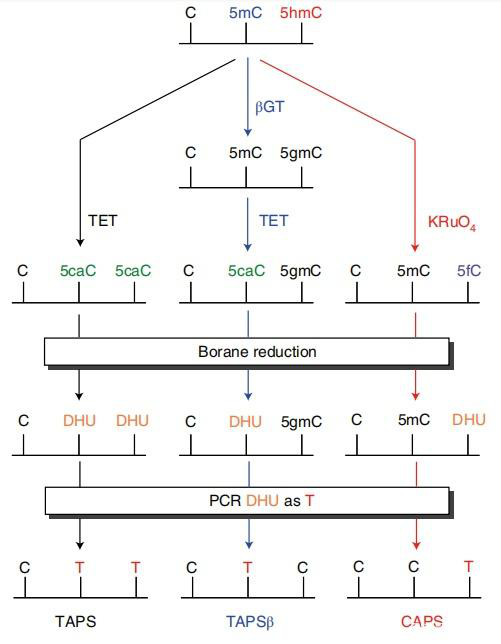
TAPS测序原理概述。图片来源:https://baijiahao.baidu.com/s?id=1735143224431472810&wfr=spider&for=pc
TAPS对ctDNA进行拷贝数变异分析
拷贝数畸变(染色体物质的大量损失或增加)被认为是癌症的一个标志,可以用于几乎所有癌症类型的非侵入性检测。对于每个cfDNA样本,作者将基因组分成1kb长的非重叠区间,计算重叠区域的高质量比对读数,并进行过滤过程以去除GC含量和映射引入的偏差。作者进一步在非癌症血浆样本上使用主成分分析来表征背景噪声,并应用了Savitzky-Golay平滑滤波器,来消除样本中实验和生物因素导致的系统误差。
作者在15/36的结直肠癌、3/8的食管癌、5/6的胰腺癌、3/5的肾癌、2/4的卵巢癌和1/2的乳腺癌血浆样本中检测到染色体畸变,灵敏度为47.5%(下图Bii),特异性为100%,涵盖了早期和晚期癌症阶段(下图 Ci)。作者还进行了ROC分析用于评估分类器的性能,结果显示ctDNA分数低至0.7%时AUC达到80%,表明即使在低ctDNA丰度下,该方法也具有较高的性能(下图D)。
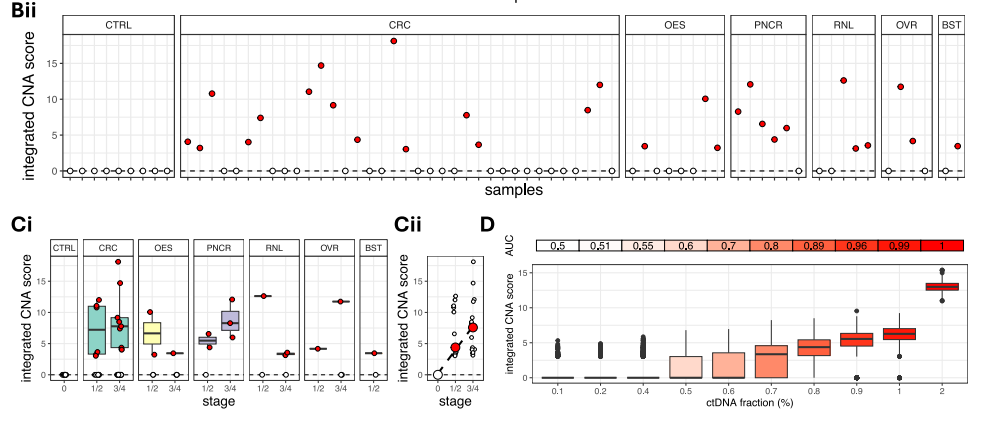
拷贝数畸变(CNA)分析。图片来源:Nature communications
ctDNA检测中的体细胞突变分析
阐明血浆cfDNA中体细胞突变的特征,主要的障碍是需要从更丰富的种系变异和测序错误中区分肿瘤起源的SNV和INDEL。为了克服这些局限性,作者采用深度(80X)WGS方法进行敏感突变检测。每个血浆样本a)与匹配的种系样本配对,以有效去除种系突变;b)使用定制软件进行处理,识别TAPS引起的变化;C)多重过滤流程去除测序误差。
结果显示,相对于非癌症对照CBS血浆样本携带平均151.1个体细胞突变,癌症血浆样本中平均含有更多的突变(425.8),并显示出更高的变异性。总体而言,此方法在17/36的结直肠癌、5/8的食道癌、5/6的胰腺癌、2/5的肾癌、1/4的卵巢癌和2/2的乳腺癌血浆样本中检测到突变负担增加的染色体臂,敏感性为52.5%(图Bii)。ROC分析显示,在ctDNA分数为1%或更高时,AUC至少为74%(图D)。
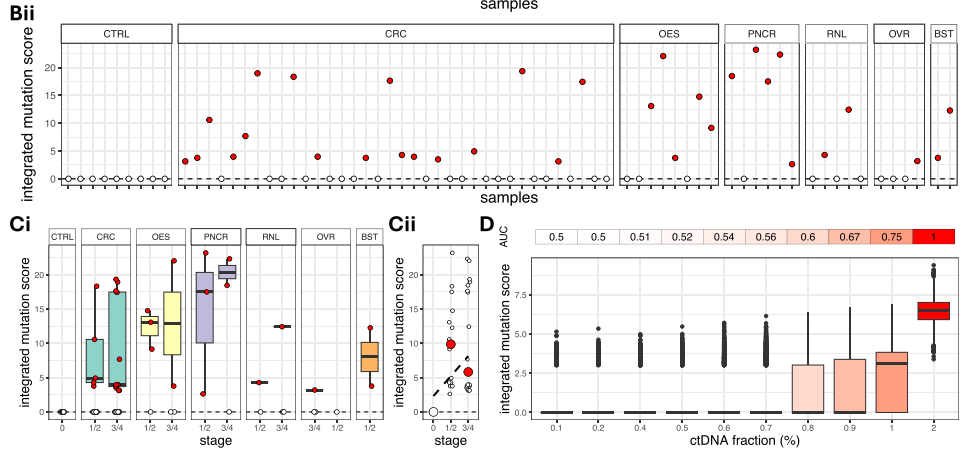
体细胞单核苷酸变异(SNV)和INDELs分析。
图片来源:Nature communications
用于ctDNA检测的甲基化信号分析
DNA甲基化异常是癌症的标志之一,可以用于疾病的诊断、治疗和监测。本研究中,作者从几个TCGA研究中提取了一组高甲基化区域,其中基线甲基化在非癌症对照中接近于零,并且只有TAPS化学转化成功才会产生阳性信号。作者在每个cfDNA样本中确定了在这些区域中至少重叠三个CPGs的片段,并计算了片段的总体甲基化水平。这种方法可在低ctDNA分数的情况下提高灵敏度。
总体而言,作者在18/36的结直肠癌、2/8的食道癌、1/6的胰腺癌、4/5的肾癌、3/4的卵巢癌和0/2的乳腺癌血浆样本中检测到甲基化负担显著较高的区域,导致45.9%的敏感性(图Aii)和100%特异性。ctDNA分数为0.9%时,AUC值为87%(图C)。
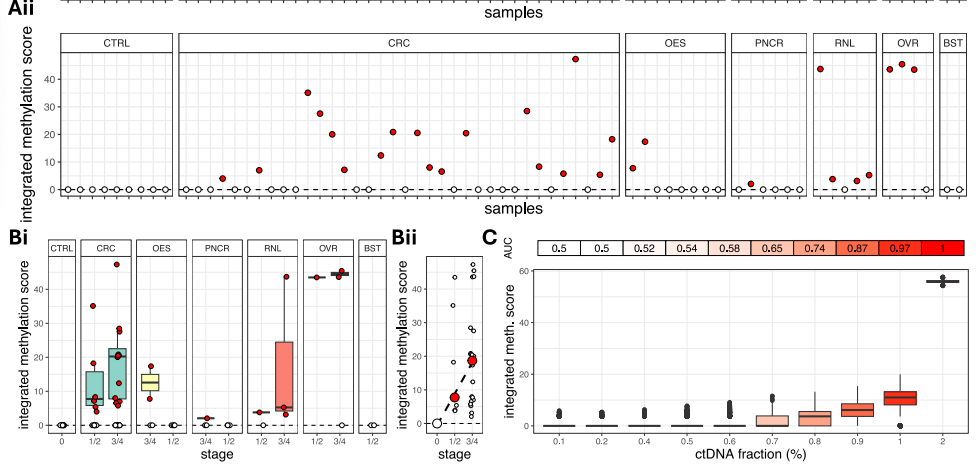
甲基化信号分析。图片来源:Nature communications
整合多种基因组数据模式用于ctDNA检测
以上分析的基因组数据模式提供了血浆中肿瘤含量的三个独立和互补的评估。作者推断,结合这些数据模式可以提高检测每个血浆样本中ctDNA的灵敏度。结果显示,异常血浆样本的多模态评分范围从1.8到36.5,而在非癌症CBS对照中,多模态评分均为零(特异性100%)。多模态结果在29/36份结直肠癌、7/8份食管癌、5/6份胰腺癌、5/5份肾癌、4/4份卵巢癌和2/2份乳腺癌血浆样本中检测到ctDNA的存在,灵敏度大幅提高至85.2%(图A)。ctDNA分数低至0.7%时AUC为86%(图C)。整合三种数据模式的多类分类器,正确识别了25/36例结直肠癌、4/8例食管癌、3/4卵巢癌、2/6胰腺癌和0/5肾癌病例的癌症起源。其总体平衡分类准确率为71.7%。
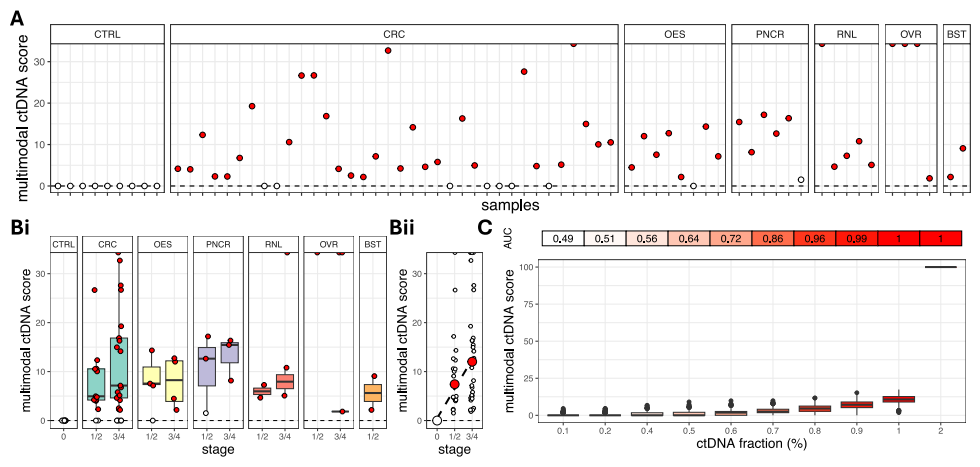
ctDNA检测的基因组数据模式整合。
图片来源:Nature communications
多模态ctDNA检测在无匹配肿瘤的结直肠癌术后MRD和辅助治疗反应跟踪中的应用
为了评估追踪疾病进展的潜力,作者进一步分析了10例结直肠癌患者的术前和术后血浆样本。结果显示,10例患者中有9例末次血浆样本中是否存在ctDNA与辅助治疗或临床结果相关。此外,在手术后或辅助治疗结束后获得血浆样本的病例中(n = 9例),无事件生存(即无复发、转移或癌前腺瘤)与治疗后检测不到ctDNA相关(图Ci, ii)。
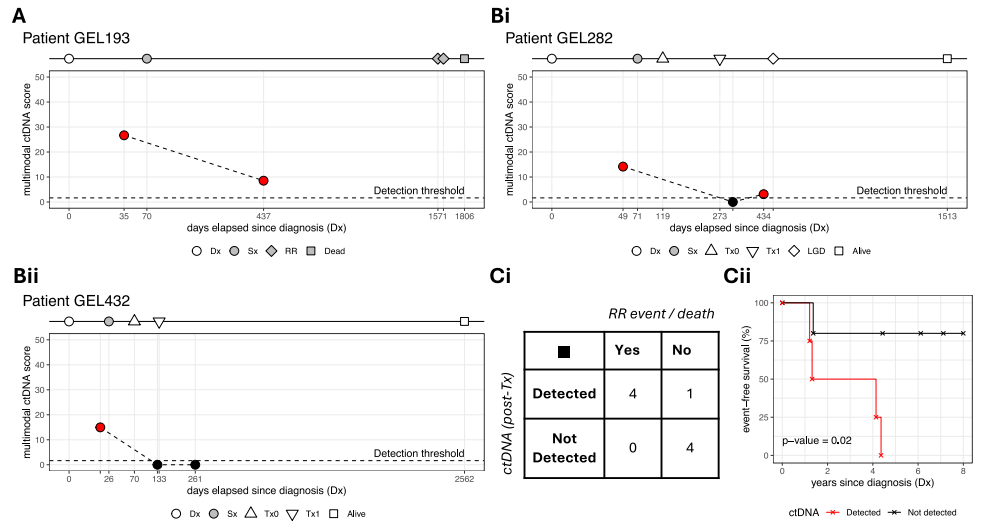
多模态ctDNA检测在无匹配肿瘤的结直肠癌术后MRD和辅助治疗反应跟踪中的应用。图片来源:Nature communications
总结与讨论
作者开发了一种使用深度(80倍)全基因组TET辅助吡啶硼烷测序(TAPS)的ctDNA检测方法,可以同时分析基因组和甲基组学数据。对有症状患者的多种癌症类型进行了诊断准确性研究,达到了94.9%的敏感性和88.8%的特异性。在ctDNA分数低至0.7%时,具有很强的辨别能力(86% AUC)。
但此方法也存在一定的局限性:甲基化特征是基于TCGA的一小部分癌症类型的数据,主要来自结直肠病例,并且使用亚硫酸测序生成。需要完整TAP特异性甲基化图谱来提高预测的准确性。此外,80倍的深度测序可能会对该方法的广泛采用构成障碍,特别是在资源有限的临床环境中。出于这个原因,未来的研究还应该集中于确定这类研究的最小可行覆盖深度。
总的来说,深度全基因组测序结合TAPS的多模态ctDNA分析方法可以高精度地检测早期和晚期癌症的癌症信号。下一步将在未选择的连续病例中进行前瞻性研究,以充分建立诊断性能。

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言














#ctDNA# #癌症诊断#
4