
【AJH】慢性髓性白血病的诊断、治疗和监测(2025更新)
2024-08-09 聊聊血液 聊聊血液 发表于上海

Elias Jabbour和Hagop Kantarjian教授近日于《American Journal of Hematology》发表综述,阐述了慢性髓性白血病的诊断、治疗和监测。
慢性髓性白血病
慢性髓性白血病 (CML) 是一种以髓系增生为主的造血干细胞恶性疾病,年发病率为2例/10万,约占新诊断成人白血病的15%,各个年龄组中均可发生,但随着年龄增长发病率逐渐增加。酪氨酸激酶抑制剂(TKI)的应用使 CML的病程彻底改观,对于绝大多数患者来说,CML已经成为一种慢性可控制的肿瘤。
MD安德森癌症中心Elias Jabbour和Hagop Kantarjian教授近日于《American Journal of Hematology》发表综述,阐述了慢性髓性白血病的诊断、治疗和监测。原文较长,现整理主要内容供参考。

概述
CML 发病机制的核心为9号染色体上的 Abelson murine leukemia (ABL1) 基因与22号染色体上的断裂点簇区 (BCR) 基因融合,从而导致癌蛋白BCR::ABL1表达,它是一种组成性活化酪氨酸激酶,通过下游信号通路如RAS、RAF、JUN激酶、MYC和信号转导和转录激活子 (STAT) 促进 CML 细胞的生长和存活。后续通过产生细胞因子无关的细胞周期和异常凋亡信号来响应细胞因子戒断,从而影响白血病的发生。
既往CML的药物治疗仅限于非特异性药物,如白消安、羟基脲和干扰素-α (IFN-α)。IFN-α治疗可抑制 Ph 阳性细胞并改善生存,但疗效不佳且毒性显著。异基因造血干细胞移植 (allo-HSCT) 可治愈,但存在致病和死亡风险。对于体能状态和器官功能良好且有适当供者的年轻患者,Allo-HSCT可作为选择。
小分子BCR::ABL1 TKI可强效干扰BCR::ABL1癌蛋白和三磷酸腺苷 (ATP) 之间的相互作用,阻断恶性克隆细胞增殖,导致CML的治疗前景发生显著变化。TKI改变了CML 的自然病史,在20年的随访中,10年生存率从约20%提高到80%-90%。虽然10年生存率约为85%,但仍有许多患者死于与CML 无关的原因(老年、第二肿瘤和其他)。伊马替尼作为一线治疗的10年 CML 特异性生存率为90%,10年 CML 耐药率仅为10%,CML特异性死亡率为10%,10年急变转化率仅5%-6%。
表现和分期
在美国,约50%诊断为 CML 的患者无症状,CML的诊断通常发生于常规体检或血液检查期间。CML可分为3期:慢性期(CP)期、加速期 (AP)、急变期 (BP),大多数 (90%-95%) 患者表现为CML-CP。CML-CP的常见体征和症状(如存在)是由贫血和脾肿大引起,包括疲劳、体重减轻、不适、易饱和左上腹饱胀或疼痛;罕见表现包括出血(与血小板计数降低和/或血小板功能障碍相关)、血栓形成(与血小板增多和/或显著白细胞增多相关)、痛风性关节炎(由尿酸水平升高引起)、阴茎异常勃起(通常伴有显著白细胞增多或血小板增多)、视网膜出血以及上消化道溃疡和出血(由嗜碱性粒细胞增多引起的组胺水平升高引起)。由于白血病细胞淤积于肺或脑血管而引起的白细胞抑制症状(呼吸困难、嗜睡、协调性丧失和精神错乱)在CP中并不常见,即使白细胞(WBC)计数超过100x109/L。脾肿大是最为统一的体征(20%-40%),肝肿大较少见(小于5%-10%)。淋巴结肿大及皮肤或其他组织浸润少见,如果存在,则更倾向于Ph 阴性 CML 或 CML 的 AP 或BP。CML转化时头痛、骨痛、关节痛、脾梗死疼痛和发热更常见。大多数患者在 BP 之前先进展为AP,但在历史上(在 TKI 可用之前)也有20%的患者在没有 AP 警告信号的情况下突然出现BP。这种可怕的突然进展为 BP 在 TKI 时代极为罕见,主要发生于 TKI 治疗的前1-2年,通常作为淋巴系BP 发生于年轻患者中。另外,在TKI治疗2年以上且完全细胞遗传学反应(CCyR)中,患者通常表现为细胞遗传学反应丧失(BCR::ABL1转录物水平增加到>1%)、血液学反应丧失和AP体征作为BP发展的前奏警告。CML-AP可能是隐匿性的或表现为贫血加重、脾肿大和器官浸润。CML-BP表现为急性白血病(髓系60%,淋系30%,巨核细胞或未分化型10%),伴有症状加重、出血、发热和感染。
诊断
典型 CML 的诊断较为简单,包括在持续不明原因白细胞增多(或偶见血小板增多)的情况下,发现存在费城 (Ph) 染色体异常、t(9;22)(q34;q11),或通过荧光原位杂交 (FISH) 或分子学检测发现 Ph相关分子BCR::ABL1异常。
FISH 分析依赖于 BCR 和 ABL1 基因特异性大基因组探针的共定位。通过 FISH 分析同时比较骨髓和血液样本,发现具有高度一致性。FISH研究根据使用的探针,假阳性范围可能为1%-5%。
逆转录酶-聚合酶链反应 (RT-PCR) 扩增 BCR 和 ABL1 之间剪接点周围的区域,在检测微小残留病方面具有高度敏感性。PCR检测可以是定性 (QPCR)(提供关于BCR::ABL1转录物存在的信息)也可以是定量(评估BCR::ABL1转录物的量)。QPCR可用于诊断CML;定量 PCR 是监测残留病变的理想方法。同时进行的外周血和骨髓 QPCR 检查显示两者高度一致。PCR可能出现假阳性和假阴性结果。假阴性结果可能是由于 RNA 质量较差或反应失败;假阳性结果可能是由于污染。根据检测程序、样品处理和实验室经验,部分样品可能出现0.5-1的对数差异。数字PCR(ddPCR) 对BCR::ABL1具有高灵敏度和特异性,在部分实验室用于检测BCR::ABL1转录物;但主要用于评价无治疗缓解 (TFR) 情况下的分子学反应 (MR)。对于相关目标和监测,无需进行重复骨髓监测,CCyR(细胞遗传学分析的0% Ph阳性中期)相当于FISH测试阴性(±2%)和国际标准(IS) BCR::ABL1转录物<1%(也称为分子学疾病减少MR2-2-log)。细胞遗传学部分反应(PCyR;ph阳性中期≤35%)相当于BCR::ABL1转录物(IS)≤10%。
Ph 染色体通常100%处于中期分裂相,多为唯一异常。10%-15%的患者有额外的染色体异常 (ACA),通常同时发生在 Ph 阳性细胞(克隆演变)中,包括8号染色体三体、17号等臂染色体、22q或double Ph 额外丢失或其他。
90%的患者有典型的t(9;22) ;5%有变异易位,可以是简单的(涉及9号染色体和2号染色体以外的染色体;即仍然是 ABL1 易位)或复合体(除9号和22号染色体外,还涉及一条或多条染色体)。Ph变体患者对治疗有反应,且预后与 Ph 阳性 CML 相似。约2%-5%的患者表现为 CML 的形态学表现,经细胞遗传学检查无 Ph 阳性。如果 FISH 和 PCR 研究证实为Ph 阳性BCR::ABL1重排CML,则此类患者对 TKI 治疗的反应和预后与 Ph 阳性 CML 患者相似。
骨髓穿刺适用于疑似 CML 的所有患者,因其可确认诊断(例如细胞遗传学分析),并提供原始细胞和嗜碱性粒细胞百分比等信息用于分期。基线细胞遗传学分析可检测ACA,特别是 i(17)(q10)-7/del7q 和3q26.2重排与相对较差的预后相关。需要进行基线逆转录酶-聚合酶链反应,以确定在评估 TKI 治疗反应时可以适当随访的特定重排类型。BCR::ABL1的典型易位可导致 e13a2 或 e14a2 转录物,产生 p210 癌蛋白。约1%的患者可能有 e1a2/a3 转录物,导致 p190 癌蛋白缩短,这些患者的预后可能更差。约2%-5%的患者携带p210 BCR::ABL1的 e13a3 或 e14a3 变体,或 e19a2 转录物 (p230)(罕见;惰性 CML 病程),通过常规探针可能产生假阴性PCR。如果在诊断时未进行检查,将导致TKI 治疗中可能处于完全 MR 的错误印象。
鉴别诊断
CML 必须与类白血病反应(leukemoid reactions)相鉴别,后者通常呈WBC 计数低于50x109/L、毒性粒细胞空泡化以及粒细胞内Döhle体,无嗜碱性粒细胞增多,白细胞碱性磷酸酶 (LAP) 水平正常或升高。临床病史和体格检查一般可提示类白血病反应的起源。皮质类固醇很少引起中性粒细胞极度增多伴左移,但这种异常是一过性的,持续时间较短。
CML 较难与其他骨髓增生或骨髓增生异常 (MDS) 综合征相鉴别。伴或不伴有骨髓纤维化的无基因髓样化生患者经常有脾肿大、中性粒细胞增多和血小板增多。伴有铁缺乏的真性红细胞增多症,血红蛋白和红细胞压积值正常,可表现为白细胞增多和血小板增多。此类患者的 LAP 评分通常正常或升高,WBC计数低于25x109/L,且无 Ph 异常。一些患者可能存在非典型 CML 临床表现,其特征为常规核型分析或 FISH 未检出 Ph 染色体,以及PCR 未检出BCR::ABL1转录。此时的鉴别诊断包括 MDS/骨髓增殖性肿瘤 (MPN) 和慢性粒单核细胞白血病,这些患者通常对 TKI 治疗无应答,与 Ph 阳性 CML 患者相比预后通常较差,中位生存期为约2-3年。二代测序可以通过检测非 ABL1 突变有助于完善诊断,从而区分这些疾病。例如,在25%的非典型 CML 病例中发现 SETBP1 突变,其中 SF3B1 突变与 MDS/MPN 伴环形铁粒幼细胞和明显血小板增多相关(MDS/MPN-RS-T;50%-70%的病例),与野生型 SF3B1 病例相比中位生存期较长(3.3年 vs 7年)。此外,慢性中性粒细胞白血病或非典型 CML 患者中存在 CSF3R 突变可能表明对芦可替尼治疗的潜在敏感性,完全缓解率为50%-60%。
最大的诊断困难在于有脾肿大和白细胞增多但没有 Ph 染色体的患者。在部分患者中,即使细胞遗传学模式正常或不典型,但仍可显示BCR::ABL1杂交基因。Ph阴性且BCR::ABL1阴性的患者视为 Ph 阴性 CML 或慢性粒单核细胞白血病。罕见情况下,患者有骨髓增生,几乎只累及中性粒细胞、嗜酸性粒细胞或嗜碱性粒细胞谱系;这些患者为慢性中性粒细胞、嗜酸性粒细胞或嗜碱性粒细胞白血病,没有 Ph 染色体或BCR::ABL1基因的证据。原发性血小板增多症可见孤立的巨核细胞增生,血小板增多明显,脾肿大。偶尔出现原发性血小板增多症临床特征(血小板增多症但无白细胞增多症)的患者可能患有CML;细胞遗传学和分子学研究显示 Ph 染色体、BCR::ABL1重排或两者均有,因此需要进行适当的诊断和治疗。
选择一线TKI治疗
CML-CP一线治疗的目的
TKI的一线治疗选择取决于以下几个考虑因素:(1) 治疗目的,(2) 患者年龄和合并症,(3) 所考虑 TKI 的成本和可负担性,(4) CML 风险特征。CML治疗的主要目的是使生存正常化并可能实现持久的DMR(MR4或MR4.5持续2-5年以上)和 TFR 状态。对于生存期延长/正常化,至少16项随机试验显示了伊马替尼和二代 TKI 的等效性,如果患者适合且依从性好,其治疗可以从伊马替尼迅速变为二代TKI。如果治疗的目的是持久的DMR/TFR,则二代 TKI 可能比伊马替尼更快达到,对于CML-CP 年轻患者(TKI 停药和 TFR 状态很重要)可考虑使用。
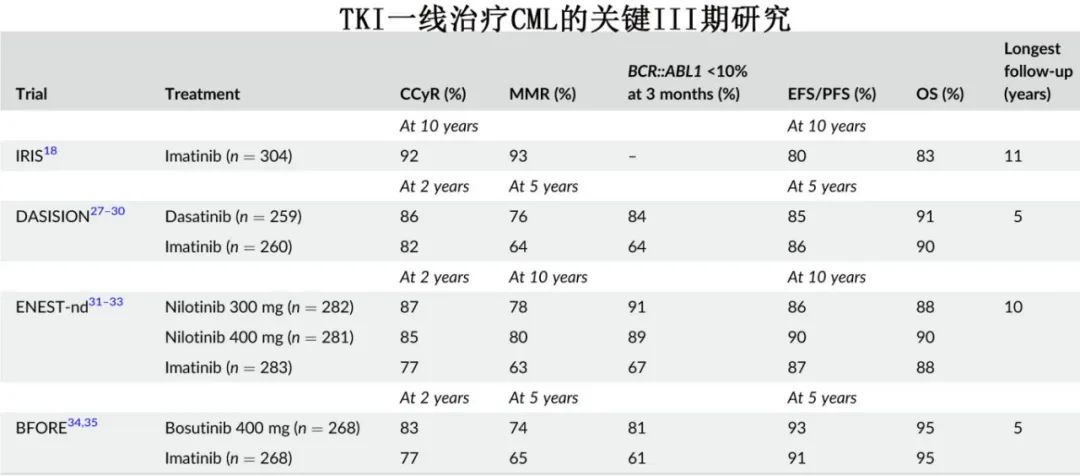
患者年龄、合并症和TKI毒性特征
虽然多种 TKI 可用于治疗新诊断的CML-CP,但在决定是否使用时均需考虑不同的毒性特征。大多数 TKI 耐受良好,可根据毒性调整剂量、充分监测和支持治疗。
伊马替尼可引起生活质量副作用,包括体重增加、疲乏、外周和眶周水肿、骨骼和肌肉疼痛、恶心和其他。但大多数为轻度至中度。长期治疗时不到5%-10%的患者发生肌酐升高。此外还报告了罕见的神经毒性(帕金森综合征恶化、痴呆样)。
对于有发生胸腔积液风险(现有肺损伤)的患者,应避免使用达沙替尼,这对于有肺部疾病(例如慢性阻塞性肺疾病)、心脏疾病(例如充血性心力衰竭)或不受控制的高血压病史的患者较为重要。肺动脉高压 (PAH) 是达沙替尼罕见但重要的并发症,既存 PAH 的患者应考虑使用其他TKI。达沙替尼也抑制血小板功能,同时服用抗凝剂的患者发生出血性并发症的风险可能增加。
尼洛替尼与高血糖症相关,糖尿病未控制的患者在开始治疗时应谨慎。糖尿病或有胰腺炎病史的患者应避免或慎用尼洛替尼。在临床前开发过程中,尼洛替尼显示可能延长 QT 间期,在药物获批后设置参数以监测该并发症。患者应在空腹状态下服用尼洛替尼,以避免药物暴露过量。尼洛替尼也与 AOE 相关,如缺血性心脏病、缺血性脑血管事件和外周动脉闭塞性疾病 (PAOD)。在 ENEST-nd 试验的10年随访中,尼洛替尼 300 mg BID 组24.8%的患者发生AOE。在有风险因素如糖尿病或冠状动脉、脑血管或外周动脉疾病的患者中,应限制或避免使用尼洛替尼。在有 AOE 病史的患者应避免使用尼洛替尼,Bosutinib和伊马替尼是与 AOE 相关的最安全TKI。
Bosutinib的副作用为胃肠道、肝脏和肾脏。在有此类合并症的患者中,应避免或慎用Bosutinib。炎症性肠病或肾功能不全患者应避免使用Bosutinib。为避免早期自限性胃肠道毒性(腹泻),建议以每日 100-200 mg 开始Bosutinib治疗1-2周,然后递增至每日200-300 mg 治疗2-8周,然后稳定在每日400 mg或最佳个体剂量。
最后,患者年龄在治疗决策中起重要作用。年龄低于50-60岁的患者预期可存活30年以上,因此,诱导持久的 DMR 可带来停药。与伊马替尼相比,二代 TKI 诱导的 DMR 更快。而持久 DMR 和潜在停药问题在老年患者中的作用较小,停药的重要性相对较低。
随着持续监测和评价,尽管罕见,但仍出现了临床重要的毒性。在2%-3%的患者中观察到肾功能不全和偶发肾衰竭(肌酐升高>2-3mg/dL),且伊马替尼和Bosutinib组的频率高于其他 TKI 组。TKI停药和/或降低剂量后趋于逆转。极少数情况下患者可发生 TKI 相关周围神经病变或中枢神经毒性,误诊为痴呆或阿尔茨海默病;这些可在 TKI 停药后逐渐逆转。
虽然大多数副作用(即使是重度)可以通过降低 TKI 剂量进行管理/逆转,但某些副作用可能是禁忌,需要改变 TKI 治疗。
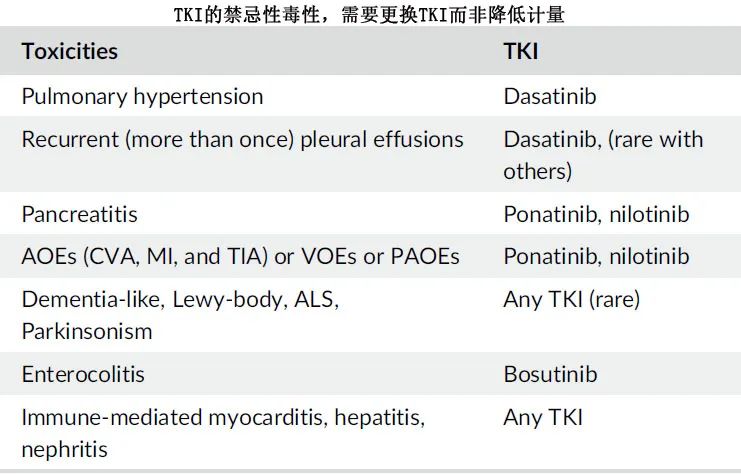
属于禁忌的毒性包括:(1) 复发性胸腔积液:达沙替尼最常报告,伊马替尼和尼洛替尼较少报告;对短疗程的类固醇有反应。(2) 血管痉挛性或VOE:包括脑血管意外、心肌梗死或不稳定型心绞痛;ponatinib和尼洛替尼更常见,伊马替尼和Bosutinib最少见。(3)PAH:达沙替尼治疗可发生(1%-2%),但其他 TKI 治疗时很少发生。TKI停药及短期类固醇和枸橼酸西地那非治疗后PAH可缓慢逆转。(4) 胰腺炎(尼洛替尼为2%,ponatinib为2%-4%)。(5)神经系统问题(痴呆样、帕金森综合征):罕见,在 TKI 停药后缓慢可逆。(6) 免疫介导事件(肺炎、心肌炎、心包炎、肝炎和肾炎):通常可通过 TKI 停药和短期高剂量类固醇(例如甲泼尼龙50 mg BID,3-5天)逆转。(6) 重度结肠炎:需要观察bosutinib停药后是否可恢复。
因为禁忌性毒性而转换 TKI 治疗时,新 TKI 的剂量并非必须是耐药时的推荐剂量。一般而言这些患者中许多具有良好的MR,新 TKI 的剂量可以更低,特别是如果患者已经缓解 (≥MR2),例如达沙替尼每日20-50 mg、bosutinib每日100-300 mg、尼洛替尼每日 200 mg 或 150 mg BID 和ponatinib每日15-30 mg。
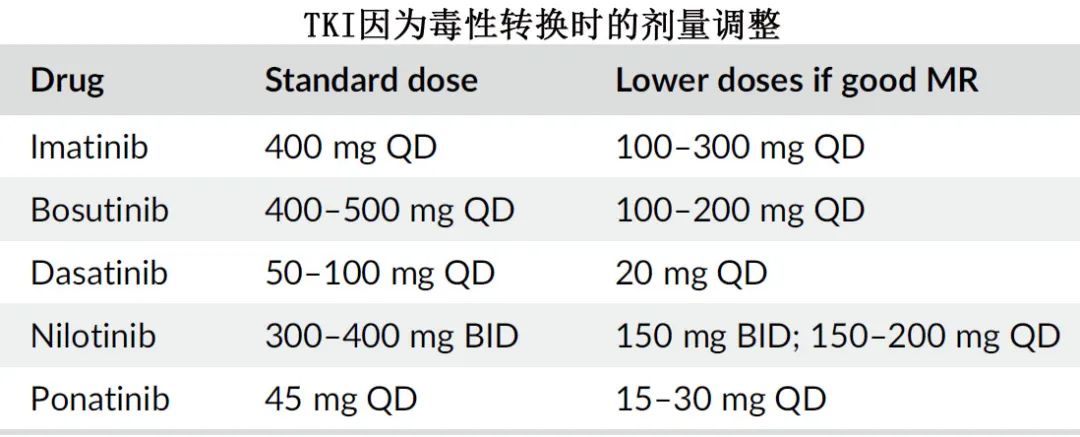
TKI 交叉不耐受可能比以前更常见。当患者对一种 TKI 不耐受/发生毒性时,也可能常对其他 TKI 不耐受。对新 TKI 的不耐受/毒性可能与既往 TKI 相同(例如在达沙替尼治疗期间发生胸腔积液的患者在bosutinib治疗期间也有发生胸腔积液的风险),亦或不同。
TKI的成本和可负担性
BCR::ABL1 TKI将 CML 从致死性癌症转化为预期寿命接近正常的慢性癌症。由于伊马替尼与二代 TKI 的长期生存期相当,以生存期为终点时,伊马替尼仿制药可能是最佳一线治疗;以TFR为终点时,二代TKI的仿制药可能是最佳一线选择。二代TKI可提供给高危患者,伊马替尼可提供给低危和中危患者。
CML-CP风险状态
对于 CML-CP 患者,多个风险评分(Sokal等或Hasford等开发)帮助预测预后。低危或中危患者预期可在伊马替尼、达沙替尼、尼洛替尼或bosutinib治疗后达到最佳缓解,而二代 TKI 作为一线治疗可能对高危患者更有益;高危CML 患者达到 CCyR 和 MMR 早期里程碑(重要时间点)的可能性更低,疾病转化为 CML-AP 或 CML-BP 的风险更高。
可选择获批用于一线 CML 治疗的任何TKI,包括伊马替尼、达沙替尼、bosutinib或尼洛替尼。虽然已证实二代 TKI 在早期替代标志物方面优于伊马替尼,但伊马替尼在大多数 CML 患者中仍高度有效。在 MD 安德森,当选择BCR::ABL1 TKI时会考虑以下问题:治疗目的(生存还是TFR);TKI成本和患者承受能力;患者年龄、合并症和 TKI AE 特征;CML风险类别。激酶结构域突变特征在选择初始 TKI 中没有作用,但在 CML 耐药背景下具有相关性。
监测治疗反应:替代终点和里程碑
由于预计接受 TKI 治疗的 CML 患者生活接近正常,因此预后的替代标志物很重要。由于技术的进步,可用于监测的侵入性检查比传统骨髓检查更少(除非更换 TKI 或特殊情况下[如非预期骨髓抑制],以排除 CML 转化或发生 MDS 综合征或其他骨髓疾病)。

监测
基线时所有患者均应接受骨髓检查以确定诊断,评估骨髓原始细胞和嗜碱性粒细胞的百分比,并进行细胞遗传学分析以确认是否存在费城染色体,并评估ACA,尤其是 i(17)(q10)-7/del7q 和3q26.2重排。开始治疗后3、6和12个月进行随访骨髓检查是研究试验的主要内容,但在常规实践中没有必要。另一种方法是使用外周血的FISH 和PCR,如果患者反应最佳,且BCR::ABL1转录物(IS) <1%,则可以省略骨髓检查。
在治疗的第一年,每3个月通过 PCR 监测外周血是合理的。一旦患者证实MMR(6个月内2-3次MMR),则每6个月监测一次BCR::ABL1转录物是充分和安全的。对于 CCyR(MR2) 患者,达到和维持 DMR 对生存期的意义值得商榷(除非治疗目标是持久 DMR 和TFR)。无论是否达到 MMR 或DMR,CCyR/MR2患者的生存期相似。
确定治疗失败的要点;ELN/NCCN指南中“警告”和“失败”信号的意义
欧洲白血病网络 (ELN) 建议和美国国家综合癌症网络 (NCCN) 指南建议并更新了定义“警告”信号和“失败”(耐药)信号的治疗里程碑,包括3个月和6个月时的“早期MR”里程碑(BCR::ABL1转录物 [IS]<10%)和后期的分子学里程碑(12+个月时MR2;36-48个月时MMR)。这些里程碑可能使社区实践混淆,导致 TKI 经常和不必要地改变。随着数据的成熟,建议/指南也可进行简化。
首先,除非有明确证据表明 CML 血液学耐药(不仅仅是BCR::ABL1转录物 [IS] >10%),否则在3个月标志之前或前后更换 TKI 是不必要的。第二,6个月BCR::ABL1转录物 (IS)>10%可能表明需要从伊马替尼更换为二代TKI,而二代 TKI 治疗时的该水平不应导致考虑三代 TKI 或allo-HSCT(除非存在 T315I 突变)。第三,基于长期随访结果,应消除ELN“警告”信号,而失败/耐药信号可以重述为“警告/谨慎”信号,因为它们预测的预后较差,但不是非常严重。第四,耐药定义为12个月或之后BCR::ABL1转录物 (IS) > 1%,是 CML 耐药和需要改变 TKI 治疗的明确指征。第五,在MMR(或DMR)患者中改变 TKI 以达到CMR(检测不到BCR::ABL1转录本)和TFR,或≥MR2的患者加深MR,是不必要的,可能造成比获益更大的伤害(新毒性、成本增加、混乱)。第六,在老年患者的特殊情况下,持续BCR::ABL1转录物 (IS) 1%-10% 达≥2年(且已用尽所有可能的安全 TKI 排列)可能不需要考虑allo-HSCT,因为这些老年患者可能在接受每日最佳 TKI 治疗(联合羟基脲、阿糖胞苷、去甲基化药物和高三尖杉酯碱)后10+年仍处于无CCyR的慢性期。
在多项研究中,TKI治疗12个月或之后达到CCyR(Ph阳性中期0%;BCR::ABL1转录物 [IS]≤1%)与达到较低程度缓解相比有显著生存获益,达到 CCyR 是 TKI 治疗的主要终点;达到BCR::ABL1转录物[IS]≤0.1%(MMR) 与无事件生存率适度改善、CCyR持续时间可能更长相关,但与生存获益无关;实现持久 DMR 可提供治疗中止和 TFR 的可能性。未达到 MMR 或 DMR 不应解释为需要改变 TKI 治疗或考虑allo-HSCT。NCCN 和 ELN关于耐药和反应不佳标准的更新随着时间的推移而发展,建立改变 TKI 治疗的保守标准。长期随访研究表明,既往符合早期反应不佳或耐药标准的患者在不改变 TKI 治疗的情况下仍具有极好的长期生存率。因此,应消除“警告”信号,而失败/耐药信号可以重述为“警告/谨慎”。与之相反,定义为BCR::ABL1转录物(IS) 在12个月或之后>1%的耐药是 CML 耐药的明确指征,需要改变 TKI 治疗。对于伊马替尼治疗2年时 BCR::ABL1转录物 (IS) 持续1%-10% 的老年患者,应采用更保守的方法,这些患者的10年 CML 特异性生存率与转录物<1%的患者相似。
TKI耐药的管理
随着TKI 的广泛使用和 CML 患病率的增加,更多的患者出现治疗耐药。如前所述,治疗10年后,15%-25%的患者发生 TKI 不耐受,这是改变 TKI 治疗的最常见原因,但如今可以通过降低相同 TKI 的剂量来安全管理。治疗10年后约10%的患者发生耐药,定义为治疗12个月后 BCR::ABL1 转录物 (IS) >1%、血液学或细胞遗传学反应丧失或 CML 转化。耐药的共同机制涉及BCR::ABL1激酶结构域的点突变,其损害现有 TKI 的活性。二代 TKI 可克服伊马替尼耐药的大多数突变,但也出现了对二代 TKI 耐药的新突变。一个重要的突变 T315I 被称为“守门员”突变,对目前可用的所有TKI均表现出耐药(ponatinib、asciminib和奥雷巴替尼除外)。
在确定TKI 耐药和转换治疗之前,应评估治疗依从性和药物相互作用。依从率越低,预后越差。年轻患者、治疗不良反应患者和需要剂量递增的患者依从率较低。
二代TKI
二线 TKI 的临床研究如下。
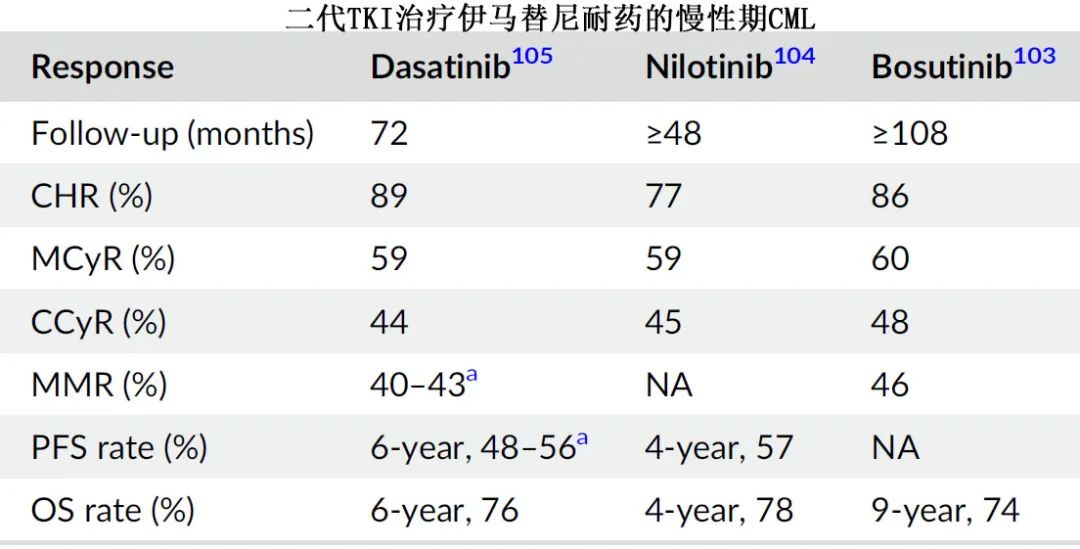
基于这些研究,出现了几个值得注意的想法。首先,达沙替尼、bosutinib或尼洛替尼二线治疗可使伊马替尼疗效不佳的患者产生较高的缓解率,包括较高的 MMR 率,且优于伊马替尼升剂量。其次,早期转换为二线 TKI 可能比晚期转换更有效。
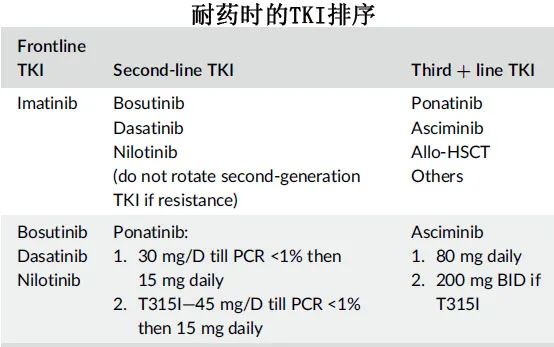
一旦患者在二线/后线治疗(甚至作为一线治疗)中对二代 TKI 产生耐药,除非存在指导(guiding)突变,否则不适合转换为另一种二代TKI(细胞遗传学/MR 率较低),换用三代 TKI 效果更好。
三代TKI
三代TKI Ponatinib和Asciminib的临床研究如下。
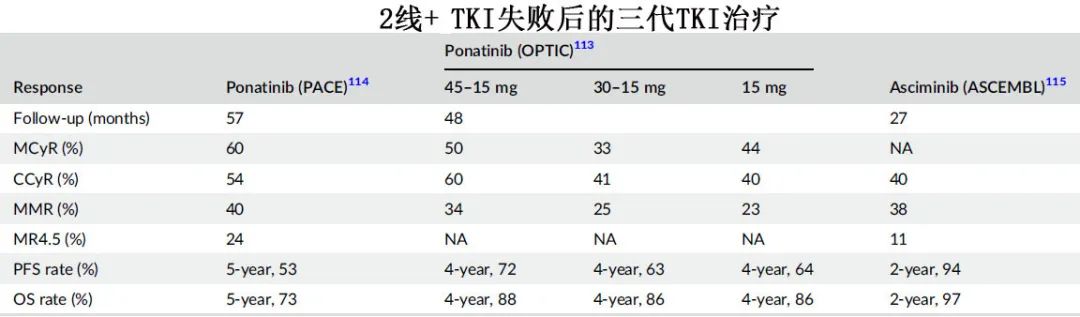
Ponatinib是三代TKI,也是第一种对 CML 伴 T315I 突变表现出疗效的TKI,其抑制BCR::ABL1的效力是伊马替尼的500倍。建议T315I突变CML使用Ponatinib 45mg每日一次,非T315I突变CML使用Ponatinib 30mg每日一次,一旦BCR::ABL1转录物(IS)≤1%则将剂量减少到15mg每日一次。
Ponatinib与二代和三代 TKI尚无头对头随机研究,但研究表明,与二代 TKI 相比,Ponatinib的效力更高;此外Ponatinib真实世界的结果优于临床试验。对于对一种二代 TKI 耐药的患者,应优先考虑将Ponatinib作为最佳TKI。下表总结了耐药患者 TKI 排序的不同情况。
阿思尼布(Asciminib)是一种STAMP抑制剂,结合BCR::ABL1蛋白的肉豆醇基位点,通过与所有其他ABL1激酶抑制剂不同的机制将BCR::ABL1锁定为非活性构象。FDA批准阿思尼布用于治疗既往接受过两种以上的TKI治疗的Ph+CML-CP成年患者,并完全批准用于治疗携带T315I突变的Ph+ CML-CP成年患者。阿思尼布在国内已申报上市,用于慢性髓细胞白血病。阿思尼布还开展了对比研究者选择的TKI治疗初治TKI的III期研究,第48周的MMR率优于研究者选择的TKI(伊马替尼、尼洛替尼、达沙替尼和Bosutinib),具有临床意义和统计学意义。
目前尚无头对头随机研究对比阿思尼布和Ponatinib,仅有倾向性评分匹配对比。
奥雷巴替尼已在中国上市,用于治疗任何TKI耐药并伴有T315I 的CP和AP CML成年患者,以及治疗对一代和二代TKI耐药和/或不耐受的CML-CP成年患者。在北美已开展并发布奥雷巴替尼用于Ponatinib和/或阿思尼布治疗后的CML-CP和Ph阳性急性淋巴细胞白血病的研究结果。
如何选择二线或三线治疗方案
在治疗失败时患者应接受骨髓检查,以确定 CML 期和ACA。所有患者均应检测 CML 细胞的BCR::ABL1激酶结构域突变,因其有助于指导 TKI 的选择。如果患者具有以下突变:Y253H、E255K/V或F359C/V,医生可能倾向于达沙替尼或bosutinib;存在 V299L 和 F317L 突变的情况下,可能倾向于尼洛替尼。对于缺乏这些突变的患者,应根据既存疾病、毒性特征和费用进行选择。
尽管 Sanger 测序 (SS) 一直是 ABL1 激酶结构域突变筛查的金标准,但二代测序 (NGS) 的使用越来越多,研究也支持在临床决策中加入基于 NGS 的 ABL1 激酶结构域突变筛查结果。ddPCR也已评估用于检测对二代 TKI 耐药的 CML 患者的 ABL1 激酶结构域突变,其非常准确和特异,并且比 NGS 更敏感,以0.5%的检测下限检测 ABL1 突变,不受BCR::ABL1水平影响;快速获得检测结果,从采样到获得结果的时间为2天。
对二代 TKI 耐药但无指导突变或 T315I 突变的患者,适用三代 TKI 并考虑allo-HSCT。
异基因造血干细胞移植
自TKI出现后,CML-CP 接受 allo-HSCT 的患者数量便显著减少,但随着 CML 患病率的增加,allo-HSCT可能再次开始上升,每年约1%-2%的患者对多种 TKI 产生耐药,因此需要allo-HSCT。当患者进展为 AP/BP 时,allo-HSCT具有更重要的作用。对于对二代 TKI 耐药(且无指导突变)或可能携带 T315I 突变的 CML-CP 患者(在泊ponatinib或阿思尼布治疗后),allo-HSCT仍是重要的治疗选择。既往暴露于 TKI 不会对移植结局产生负面影响;如果移植患者在 MR 状态更好(CML负荷更低)的情况下接受allo-HSCT,则预后可能更好。
应考虑allo-HSCT成本与 TKI 成本和可用性。Allo-HSCT是一次性、治愈性手段。鉴于现有TKI仿制品的长期获益和成本,一线治疗中不应进行allo-HSCT,而在挽救治疗中可以根据地区差异、治疗成本和二代 TKI 的可用性讨论 allo-HSCT 与二代/三代 TKI 的价值。
什么情况下不应将 allo-HSCT 视为 TKI失败/二代 TKI 耐药后的选择?随着经验的积累,“患者必须达到 CCyR 或考虑进行allo-HSCT”的教条在老年患者(例如年龄>65-70岁)中可能已不那么适宜。这些患者可以选择放弃治愈性allo-HSCT,从而在继续 TKI 治疗而未CCyR 的情况下获得相当长且接近正常的寿命。这些患者可通过单独接受最佳 TKI 治疗维持慢性期,或联合其他药物维持血液学反应伴或不伴细胞遗传学反应。
治疗持续时间和停药
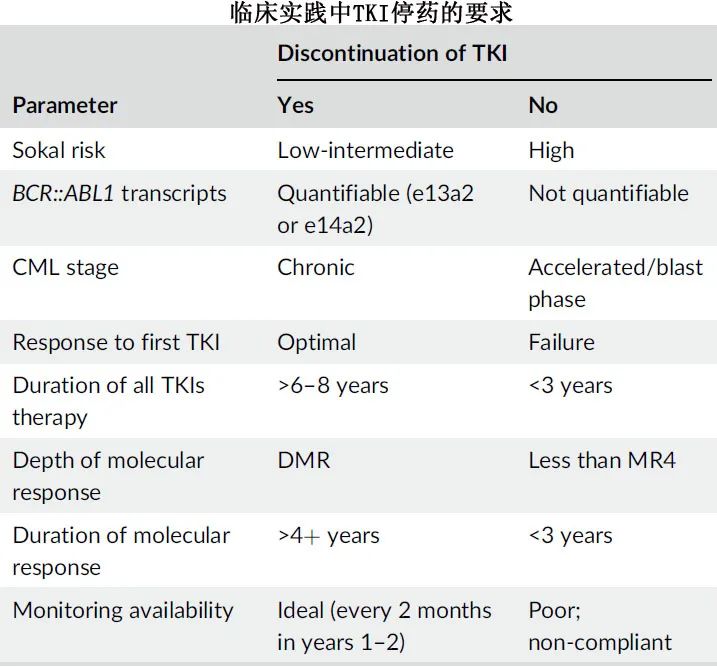
多项研究已经评估了持续DMR 的患者是否可以安全地停用TKI。一般而言,在持续DMR 2+年后停用 TKI 可获得40%-50%的 TFR 率,且更久的 DMR 可显著提高 TFR 率。例如在 EURO-SKI 试验中,DMR 每增加一年,则TFR率增加3%。MR4持续时间≥5年与较低的 MMR 丧失率独立相关。研究显示,在中止治疗的患者中,MR4范围内的单次RT-PCR 波动不会对 TFR 成功率产生负面影响,TKI停药后的分子监测频率(停止治疗后每4周一次vs.每6-8周一次)对 TFR 率无影响。据报告,在 TKI 停药前的确定持续时间内(例如1年)考虑 TKI 剂量递减的策略是可行的。
为了加深 MR 并提高 TFR 率,对多种联合治疗策略进行了评价,尤其是 TKI 联合干扰素。
持久 DMR 患者的 TKI 停药研究证明停止 TKI 治疗是可行的,并且在相当大比例的患者中TFR 状态可行。在 MD 安德森,可定量转录物的慢性期患者可中止治疗,患者需要已接受 TKI 治疗至少6-7年,已达到持久 DMR 至少4-5年,可进行密切随访。TKI停药后,建议在前12个月内每6-8周进行一次密切的分子监测,在接下来的2年内每2-3个月进行一次,此后每4-6个月进行一次。
晚期CML
CML-AP 或 CML-BP 患者可接受TKI(二代TKI,如达沙替尼或ponatinib,优于伊马替尼)初始治疗,以降低 CML 负荷,并考虑早期allo-HSCT。TKI联合化疗的缓解率在非淋系 CML-BP 中为40%,淋系 CML-BP 中为70%-80%,中位生存时间分别为6-12和12-24个月。在化疗的基础上加用 TKI 可改善CML-BP 的缓解率并延长生存期,且TKI联合强化化疗后 HSCT 的结局似乎最佳。
目前allo-HSCT是CML-CP后进展的 CML-AP(非原发性CML-AP)或 CML-BP 的唯一治愈手段,报告的长期生存率分别为15%-40%和10%-20%。以细胞遗传学克隆演变为唯一 AP 标准的患者长期无事件生存率约为60%。
WHO近期建议消除 CML-AP的定义,而将其归类为高危CML-CP,并仅定义两个 CML 阶段:CML-CP和CML-BP(原始细胞20%+)。作者认为这是不明智的:CML-AP患者,无论是新发还是演变,预后均更差,治疗有所不同,并需要对新的研究策略进行单独分析。与 CML-CP 演变而来的 CML-AP 相比,一线 TKI 治疗原发性 CML-AP 的预后更好;后者8年生存率为60%-80%,而前者的中位生存期不足3年。此外,尽管原发性CML-AP 的预后有所改善,但仍劣于CML-CP,即使是高危CML-CP。
虽然存在 ACA是 CML-AP 的标准之一,但一种特殊的细胞遗传学分子异常,即3q26易位(或MECOM),与极差预后相关,其存在应导致立即考虑allo-HSCT。
多种研究调查了基因改变的影响,值得注意的是非 ABL1 激酶结构域突变对 CML 进展风险的影响。携带体细胞变异(特别是 ASXL1 突变)的 CML-CP 患者可能需要密切监测,可能从更强效 TKI 或联合治疗中获益,也可能无法获益。这些患者可以考虑allo-HSCT。还需要更大规模的研究来更精确地评估这类突变的作用及其在 CML 中的影响。
根据 TKI 治疗的反应,AP患者应早期考虑Allo-HSCT。BP患者的唯一治愈选择也是allo-HSCT,TKI联合化疗应作为 allo-HSCT 的桥接。在 MD 安德森,CML-BP患者接受化疗(类型取决于免疫表型)和ponatinib联合治疗,一旦达到完全缓解则allo-HSCT,然后在 allo-HSCT 后给予 TKI 维持治疗。如果在治疗6个月内达到最佳缓解(CCyR;BCR::ABL1转录物[IS] < 1%),则原发性CML-AP 患者无限期接受一线二代 TKI 治疗。所有CML-AP其他患者均接受二代/三代 TKI 治疗,随后接受allo-HSCT。
参考文献
Jabbour E, Kantarjian H.Chronic myeloid leukemia: 2025 update on diagnosis, therapy, and monitoring.Am J Hematol . 2024 Aug 2. doi: 10.1002/ajh.27443.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言














#慢性髓性白血病# #CML#
64