
肺泡蛋白沉积症(Pulmonary Alveolar Proteinosis,简称PAP)是一种罕见的间质性肺病,其特征是肺泡内异常沉积表面活性物质。根据病因不同,PAP可以分为原发性、继发性和先天性三种类型。其中,自身免疫性PAP(Autoimmune PAP,APAP)是最常见的形式,占所有病例的85%以上。尽管有研究报道了PAP的一些临床表现和结局,但关于其异质性、预后及其死亡原因的研究仍较为有限。
国内研究团队通过汇总已发表的病例系列,分析了3278名PAP患者的数据,利用K均值聚类对患者进行分类,并采用逻辑回归模型识别影响预后的因素。此外,还通过对相关基因的富集分析来描述PAP的遗传机制。
研究样本来源来自295项研究的3278名患者,其中88.6%被诊断为特发性PAP(Idiopathic PAP,IPAP)。日本患者数量最多,占总人数的36.46%,其次是美国和中国。大多数患者年龄在41至50岁之间。主要通过支气管镜检查或肺活检进行诊断。
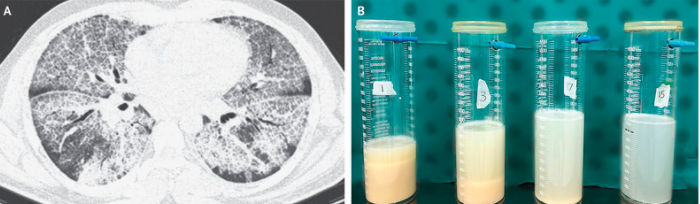
研究纳入患者主要表现为呼吸困难(49.3%)和咳嗽(35.6%),部分患者可能无明显症状。对于IPAP患者,最常见的并发症是肺部感染(尤其是由诺卡氏菌引起的细菌感染),其次是呼吸系统疾病和内分泌紊乱。全肺灌洗(Whole Lung Lavage, WLL)和粒细胞-巨噬细胞集落刺激因子(GM-CSF)疗法是常用的治疗方法。
研究共有2696名患者进行了为期五年的随访,总体死亡率为9.5%。呼吸衰竭(45.8%)和肺部感染(18.3%)是导致死亡的主要原因。根据症状的不同,患者被分为三类:无症状组、单一症状组和多症状组。结果显示,具有多个症状的患者的死亡率显著高于仅有单个或无症状的患者(P<0.05)。WLL和GM-CSF疗法显著降低了患者的死亡风险。特别是有效GM-CSF治疗的患者,其死亡风险仅为0.08。研究发现134个与PAP相关的基因,强调了免疫反应和脂质代谢在疾病发展中的作用。
此次研究首次系统地评估了PAP临床表型对其预后和死亡率的影响。研究指出,PAP的临床表现具有显著的异质性,且存在多种潜在的遗传机制。呼吸衰竭和肺部感染是PAP患者的主要死因。从严重到轻微甚至无症状的表现都有可能出现,这使得疾病的早期诊断变得复杂。全肺灌洗和GM-CSF疗法能够显著改善患者的生存率,特别是在非继发性PAP患者中。
这项全球性的PAP研究不仅提供了关于疾病临床表型、预后因素及遗传机制的全面信息,还揭示了脂质代谢途径作为潜在治疗靶点的可能性。未来的研究应进一步探索PAP的分子机制,并开发更加个性化的治疗策略,以提高患者的长期生存率和生活质量。
PAP作为一种罕见病,其诊断和治疗面临诸多挑战。首先,由于其症状多样且不典型,容易与其他呼吸系统疾病混淆,导致误诊或延误治疗。其次,虽然现有的治疗方法如全肺灌洗和GM-CSF疗法已被证明有效,但对于某些特定亚群的患者,疗效仍有待进一步验证。因此,未来的临床实践需要更多关注个体化治疗方案的设计,并结合最新的基因研究成果,为患者提供更为精准的医疗支持。
此外,随着大数据和人工智能技术的发展,如何利用这些先进技术更好地理解和管理PAP也成为了一个值得探讨的方向。例如,通过构建基于机器学习的预测模型,可以帮助医生更早地识别高危患者,并制定个性化的干预措施。同时,加强国际合作和数据共享,也有助于推动PAP研究的进步,最终造福广大患者群体。
参考文献:
Huang, J., Xie, S., Gao, Y. et al. Phenotypic heterogeneity in mortality and prognosis of pulmonary alveolar proteinosis: a large-scale, global pooled analysis of individual-level data. Orphanet J Rare Dis 20, 102 (2025).

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








签到学习
4
#肺泡蛋白沉积症#
7