
Nat Commun:新AI模型可直接“看到”肿瘤活检图像中的基因表达,涵盖16种癌症类型
19小时前 测序中国 测序中国 发表于陕西省

SEQUOIA为大规模推断及分析基因表达模式提供了一种颇具成本效益的方法,有望应用于研究和临床。
癌症是一种动态疾病,其特征是复杂的分子和细胞进化;在进化的过程中,癌症变得更加异质,分为患者间异质性和肿瘤内异质性。因此,系统地了解癌症的异质性对有效的诊断和治疗至关重要。目前,RNA-seq的进步使得在整体组织水平和空间分辨区域水平上对基因表达图谱进行全面检测成为可能,但将基因表达分析纳入临床实践仍是一个挑战。
近年来,随着组织病理学切片逐渐数字化为全切片图像(WSI),人们也开发了相关的机器和深度学习方法,从WSI中提取与分子特性相关的隐藏形态学特征。WSI的尺寸和分辨率巨大,现有模型多数基于其被裁剪后较小的“切片”开发,无法捕捉图像中多个切片之间的上下关联和层次关系。此外,由于模型复杂度高和数据集规模有限,将最先进的方法应用于WSI仍极具挑战性,如多层感知器、Transformer。
为解决上述难题,美国斯坦福大学医学院的研究人员在Nature Communications发表了题为“Digital profiling of gene expression from histology images with linearized attention”的文章,介绍了一种基于线性化Transformer的深度学习模型SEQUOIA(Slide-based Expression Quantification using Linearized Attention),可以从WSI中预测癌症转录组学图谱。研究团队利用人工智能(AI)从来自16种癌症类型的7,584个肿瘤样本中创建了SEQUOIA;SEQUOIA能够准确预测与关键癌症过程相关的基因的表达水平,可根据复发风险对乳腺癌患者进行分层,解析局部区域的空间基因表达模式。总之,SEQUOIA为大规模推断及分析基因表达模式提供了一种颇具成本效益的方法,有望应用于研究和临床。

文章发表在Nature Communications
“先前研究表明,组织病理学图像与基因RNA变异相关,”文章通讯共同作者Olivier Gevaert博士表示,“这启发我们开发一个AI模型,能否基于最新的技术进一步利用这种相关性,开发一个可以预测人体所有组织中所有基因的模型。
研究团队使用癌症基因组图谱(TCGA)中7,584个癌症活检样本的WSI和相匹配的大量RNA-seq基因表达数据开发和训练了SEQUOIA模型,共涉及16种癌症类型包括膀胱尿路上皮癌 (BLCA)、乳腺浸润癌 (BRCA)、甲状腺癌 (THCA)、肾透明细胞癌(KIRC)和肾乳头状细胞癌(KIRP)等。
接下来,通过将上述数据以及成千上万个健康细胞图像在内的其他数据集整合到SEQUOIA中,研究团队验证了该AI模型性能。结果显示,在16种癌症的20,820个基因中,SEQUOIA平均准确预测了15,344个(74%)基因;预测结果良好的基因数量与每种癌症的可用训练样本数量呈正相关。
BRCA中预测准确的基因数量最多(18,878个),同时也是可用切片最多的癌症类型(1,130张);其次是THCA(517张切片)和KIRC(514张切片),分别有18,758个和 17,623个预测准确的基因。相比之下,前列腺腺癌(PRAD)中预测准确的基因数量最少(9,535个),切片数量也最少(202张)。上述结果表明,SEQUOIA模型性能与癌症可用数据集大小(即切片数量)呈正相关。
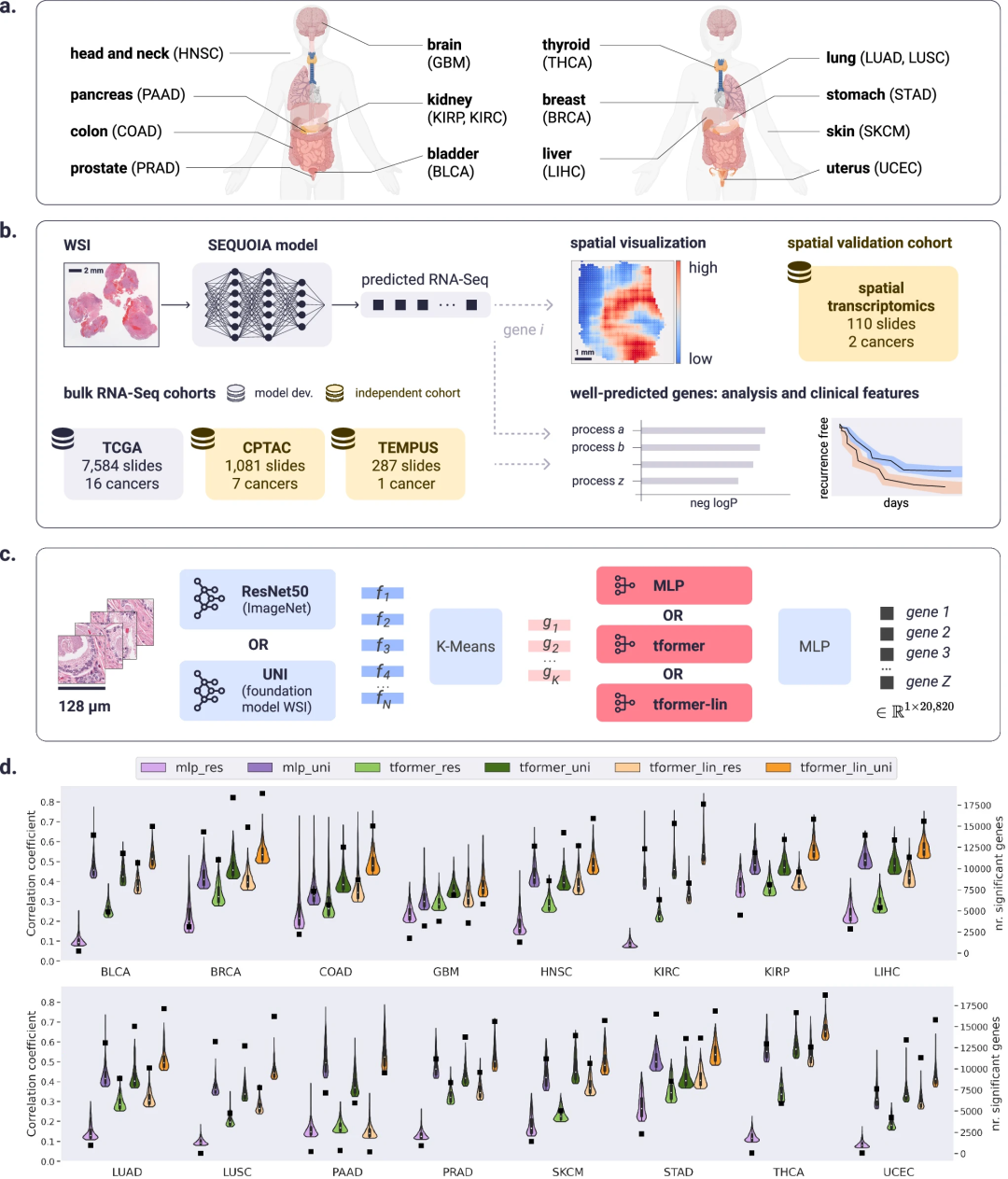
图1. SEQUOIA模型的工作流程概述
为测试SEQUOIA的泛化能力,研究团队将该模型应用于临床蛋白质组肿瘤分析联盟(CPTAC)队列6种组织的7种匹配癌症类型中。结果显示,相较其他模型组合,在7种癌症类型中SEQUOIA的相关系数更高,为0.503;预测准确的基因数量也更多,平均验证了7,159个基因;在相关系数和均方根误差方面也显著优于其他模型。这些结果表明SEQUOIA的泛化能力较强,适用于不同队列。
此外,为确定SEQUOIA模型中预测准确的基因的生物学功能,研究团队还进行了基因组分析。结果显示,预测准确的基因在几种常见的癌症类型通路中富集,包括T细胞活化、细胞-基质粘附、上皮-间质转化和氧化应激反应,这表明SEQUOIA预测准确的基因主要且特异性地与癌症发生和进展的调控有关。此外,研究团队还确定了几种预测良好的细胞类型标记,包括内皮细胞(CD69, CD93)、 CD4 T细胞(CD3E, CD4, CD48)等,体现了SEQUOIA在捕获肿瘤微环境特征方面的能力。
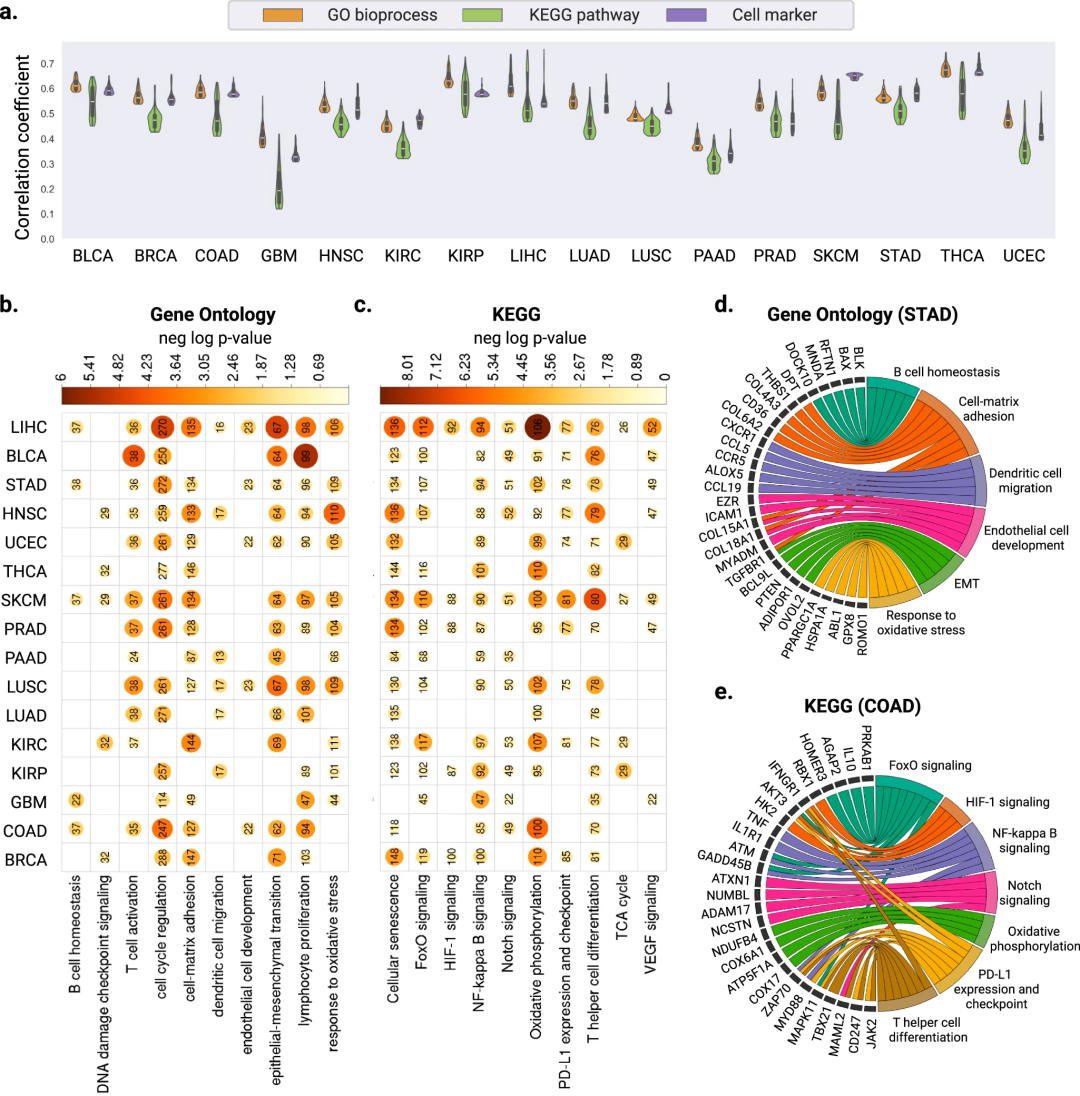
图2. 通路水平基因表达预测评估
为评估SEQUOIA在临床决策中的实用性,研究团队确定了一个包含272个基因的基因表达特征,这些基因与复发显著相关。类似的基因特征已经在商业乳腺癌基因组检测中应用,例如FDA批准的MammaPrint检测,该检测可分析70个乳腺癌相关基因的水平,为患者提供评分以确定其癌症复发的风险。
进一步,研究团队该模型应用于发现队列TCGA和两个验证队列SCANB、METABRIC中,以证明其在风险分层方面的表现。结果显示,根据SEQUOIA风险评分可将来自三个独立队列的乳腺癌患者分为高风险和低风险两个亚组;与低风险评分的患者相比,高风险评分的患者无病生存期明显更短。这表明SEQUOIA具有通过基因表达预测预测乳腺癌复发的潜力。
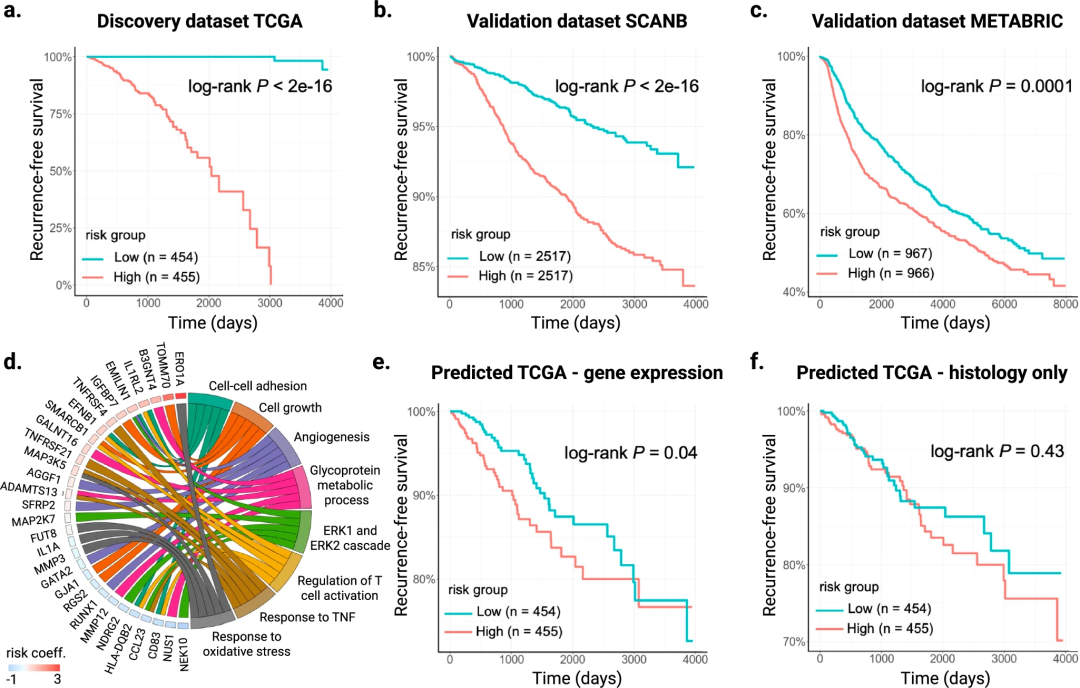
图3. 用于预测乳腺癌复发的数字基因表达特征的开发和验证
为使数据易于获取和解读,研究团队对SEQUOIA进行了编程,将基因研究结果显示为肿瘤活检的可视化地图,让科学家和临床医生可以直观看到基因变异在不同肿瘤区域的不同表现;并利用独立GBM和乳腺癌患者队列的空间转录组数据集进行性能评估。结果显示,SEQUOIA为每个基因生成了空间热图以表明其在整个玻片上的表达值,并准确预测许多基因的空间表达。
相比能够可视化活检切片中基因表达的其他工具,SEQUOIA最大的不同之处是集成了数字病理学基础模型,这些模型是在数百万张组织图像上训练出来的大型模型,与GPT、LLAMA和Gemini等流行模型类似。上述结果证明了SEQUOIA在解析异质性肿瘤组织内的空间细胞结构方面的潜力。
为证明SEQUOIA在其他癌症类型中的空间预测能力,研究团队还开发了一个用户友好的交互式Web应用程序 (https://sequoia.stanford.edu),用户可以在其中探索 TCGA队列中预测基因的空间热图。
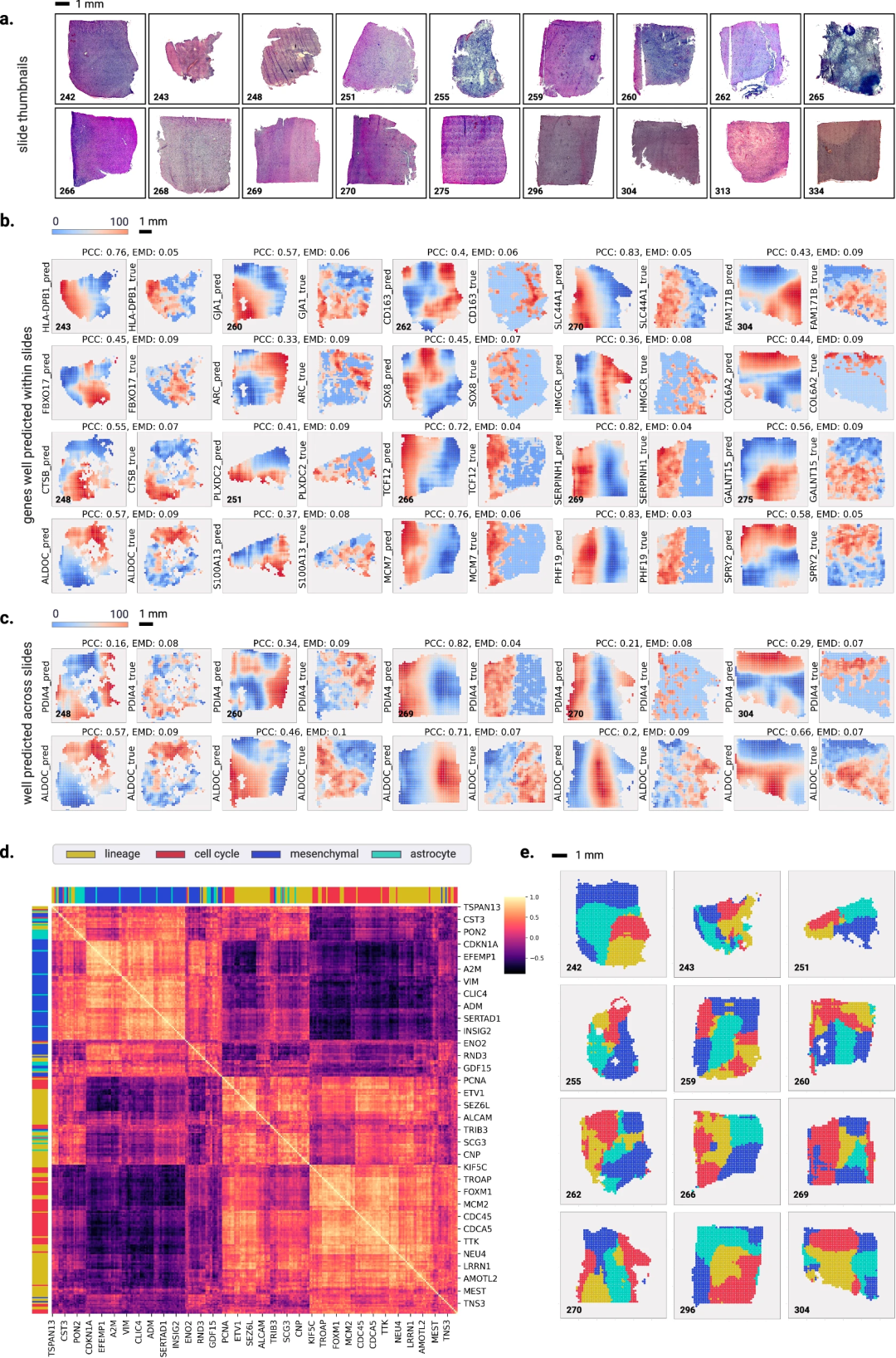
图4. 切片水平预测的基因表达空间可视化
综上所述,研究团队提出了SEQUOIA,一种用于预测WSI中RNA-seq基因表达数据的深度学习模型。通过将算法进步与对生物学功能、临床相关性和泛化能力的全面分析相结合,该研究证明了SEQUOIA在预测临床相关基因表达模式方面的价值,可为个性化癌症管理开辟道路。
由于未获得FDA批准,目前该AI模型还不能在临床环境中使用。研究团队下一步计划将SEQUOIA部署到临床工作中,并确定它在哪个阶段对医生最有益。Gevaert强调,该工具不仅限于乳腺癌。“使用我们的模型可以预测任何癌症类型的任何基因特征,除运行模型的成本外,几乎没有额外的成本。
参考文献:
1.Pizurica, M., Zheng, Y., Carrillo-Perez, F. et al. Digital profiling of gene expression from histology images with linearized attention. Nat Commun 15, 9886 (2024).
https://doi.org/10.1038/s41467-024-54182-5
2.Novel AI Tool “Sees” Gene Expression in Tumor Biopsy Images
https://www.insideprecisionmedicine.com/topics/oncology/novel-ai-tool-sees-gene-expression-in-tumor-biopsy-images/?__hstc=126276562.532115043888ec08099ea6c859d5611b.1724816722074.1732069472313.1732155706591.19&__hssc=126276562.1.1732155706591&__hsfp=2044848510&_ga=2.137523696.1157139962.1731918834-878096776.1721121866
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言













#癌症# #SEQUOIA#
4