
病例报告|成年型Sifrim-Hitz-Weiss综合征1例
昨天 中国神经精神疾病杂志 中国神经精神疾病杂志 发表于陕西省

本例是目前中国报道的第1例成年型SIHIWES。
摘 要 报道1例Sifrim-Hitz-Weiss综合征(Sifrim-Hitz-Weiss syndrome, SIHIWES)患者。患者为35岁男性,出生后有发育迟缓、肌肉萎缩、骨骼畸形、隐睾等症状,有宽前额、方形脸、低位耳、杯状耳、眼睑下垂等特殊面容。全外显子组测序技术发现CHD4基因一处杂合变异NM_001273:c.3047A>G(chr12-6701125)(p.K1016R),该突变为临床意义未明变异,变异位点未见报道,结合其症状体征诊断为SIHIWES。本例是目前中国报道的第1例成年型SIHIWES。
关键词
Sifrim-Hitz-Weiss综合征;CHD4神经发育障碍;CHD4基因;发育迟缓;全外显子组测序技术;常染色体显性遗传病
Sifrim-Hitz-Weiss综合征(Sifrim-Hitz-Weiss syndrome, SIHIWES)是一种由染色质域解旋酶DNA结合蛋白4(chromodomain helicase DNA-binding protein 4, CHD4)中的杂合错义变异引起的多系统神经发育障碍疾病,又称CHD4神经发育障碍(CHD4 neurodevelopmental disorder),呈常染色体显性遗传,2016年由SIFRIM、WEISS等发现并定义[1-2]。该综合征表现为全方面的发育迟缓,主要临床表现有轻中度智力障碍、身材矮小、特殊面容等[2-3]。目前中国并未见成年型SIHIWES文献报道,本文报告1例如下。
1 临床资料
患者,男,35岁,大专学历,因“身材瘦弱伴肢体无力30余年”入院。患者诉30余年前开始无明显诱因下出现四肢无力,后逐渐加重伴肌肉萎缩。自幼身材矮小,生长发育较同龄人迟缓,与人交流时偶有语塞现象,言语理解能力可。无反应迟钝,无明显智力异常,无吞咽困难等。平素体质一般。既往15岁因“肢体抽搐、畏光畏声”至外院就诊,查脑地形图后诊断为“癫痫”,口服德巴金1年后自行停药,后未再发作;其余既往史无特殊。其母亲、外公均有行走不利病史,母亲身材矮小,于65岁过世,外公约于50岁过世,具体原因不详。
一般生命体征检查正常。体型消瘦矮小,可见宽额、宽眼、方形下颌、双耳较小、低耳位、杯状耳等特殊面容(表1);皮肤黏膜无黄染、出血等,心、肺、腹查体无特殊。神经系统专科检查:神清语利,脑神经检查未见异常;四肢肌容量减少,关节过伸;四肢肌力4级;肱二头肌腱反射(++),肱三头肌腱反射(++),双膝反射(+++);双上肢肌张力减退;感觉系统未见异常;双侧跟膝胫试验稳准,闭目难立征阴性;双侧病理征阴性;脑膜刺激征阴性。
Tab.1 SIHIWES Clinical Phenotype Comparison表1 SIHIWES临床表型对照

血常规、生化常规、性激素检验等未见特殊异常。胸部CT示脊柱侧弯畸形(图1)。肺功能见中重度混合性通气功能障碍,弥散功能轻度下降,气道阻力正常。头颅MRI可见枕骨大孔水平脊髓变细,颅底凹陷(图1)。眼科专科检查见眼底常瞳下视网膜平伏,黄斑区色素紊乱,眼球运动无异常。心脏彩超、肌电图未见特殊异常。全外显子组测序结果表明CHD4中存在杂合变异:NM_001273:c.3047A>G(chr12-6701125)(p.K1016R)(图2),正常人变异数据库未见报道(SP2_Supporting),按照美国医学遗传学与基因组学学会(ACMG)《遗传变异分类标准与指南》评级为临床意义未名变异,此变异位点此前未见报道。
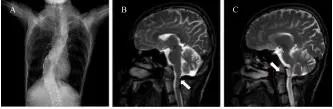
Fig.1 Imaging of the patient图1 影像学检查 A,胸部CT可见患者脊柱侧弯;B,头颅MRI T2WI序列可见患者枕骨大孔水平脊髓稍细,前后径约4 mm;C,头颅MRI T2WI序列可见患者枢椎齿状突超过钱氏线约10 mm;异常位置由白色箭头指出。
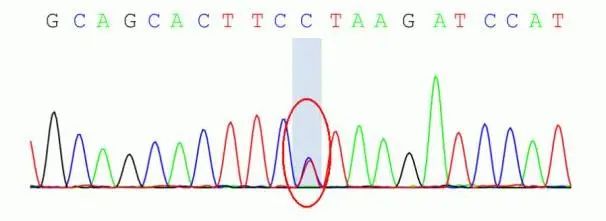
Fig.2 Genetic test results图2 基因检测结果 CHD4中存在杂合变异:NM_001273:c.3047A>G(chr12-6701125)(p.K1016R)。
目前SIHIWES尚无特殊治疗方案,大多以多学科支持治疗为主,考虑患者脊柱侧弯严重,建议进一步康复科、骨科就诊,出院后注意定期复查,避免产生肺部感染等严重并发症。1年后对患者进行电话随访,其未诉特殊不适。
2 讨论
SIHIWES是由于CHD4基因的致病性变异导致,CHD4是一种染色体结构域解旋酶DNA结合蛋白,是核小体重构和组蛋白去乙酰化酶(nucleosome remodeling and deacetylase,NuRD)抑制复合物的核心成分[4]。该复合体在人体组织内广泛表达,在调节基因转录、基因组完整性和细胞周期方面起重要作用,其活性的改变可以导致发育缺陷,癌变和加速衰老[5],这也可以解释SIHIWES涉及多系统的病变。
本文报道患者通过全外显子组测序结果表明CHD4基因中存在杂合变异,NM_001273:c.3047A>G(chr12-6701125)(p.K1016R)(图2),根据ACMG指南评级为临床意义未名变异。结合患者母亲、祖父身材矮小的家族史,我们怀疑其受到母亲的影响,希望进一步行家系分析,患者及其父亲拒绝。WEISS等[1]、SIFRIM等[2]认为鉴定意义不明的杂合CHD4 变异并不能排除SIHIWES诊断。我们对该患者与已报道的SIHIWES临床表型进行比较,结果见高度重复性(表1),该患者可诊断为SIHIWES。
截至目前,全球已有报道的SIHIWES患者33例,其中32例由WEISS等[1]在2016—2020年进行的一项多中心随访中发现并报道,1例是由周新龙等[6]报道。已报道患者年龄在新生儿至30岁之间,平均年龄为10岁,本例为SIHIWES的临床认识增加了1例35岁时被诊断的成人型患者的临床特点及基因型[3]。对比已发表病例,我们发现本例患者具有Sifrim-Hitz-Weiss综合征常见的发育迟缓、特殊面容、运动能力差等特征。以往报道的病例中15/20有眼科相关问题[3,6],如斜视、散光和屈光不正,本例患者眼底检查发现黄斑区色素紊乱,这与周新龙等报道的1例2岁的女性患儿有同样的发现,这提示在SIHIWES患者的临床监测中眼底检查在眼科检查中同样有重要地位。在之前的文献中有肌无力、骨骼肌肉发育异常的报道,其中1名患者21岁时由于颈椎不稳定和长期气管切开术的并发症死亡,但无相关的详细描述和监测指标[3],结合本文报道患者患有严重的脊柱侧弯,肺功能检测示中重度混合性通气功能障碍考虑与之相关,我们考虑严重的脊柱侧弯由躯干肌萎缩、肌无力造成,这可能会导致呼吸功能异常、严重的感染等。这提示,在SIHIWES的临床工作中对于肌肉容积、骨骼发育等监测和及时干预的必要性。
患者15岁时因肢体抽搐、畏光畏声于外院查脑电图诊断为癫痫(报告已遗失),服药1年后自行停药,未再发作。此前的报道中虽有胼胝体发育不良等脑发育异常,但尚未有脑部异常放电等表现的报道。CHD4作为心脏、大脑共表达的基因,被认为是癫痫及心律失常共病的遗传学基础[7]。心律失常是癫痫意外猝死的原因之一,先前已有很多报道证明CHD4突变与先天性心脏病的相关性[3,8-9]。LIU 等[7]通过ClinGen临床有效性框架评估得出CHD4可能是癫痫的致病基因之一。虽癫痫在此前的SIHIWES病例中未有提及,但仍是临床上需关注的重点。对于怀疑遗传性疾病合并癫痫病史的患者,不能忽视CHD4突变相关疾病的可能性,这对避免癫痫严重合并症的发生,提高患者生存期及生活质量起到重要作用。
综上,本案例对临床工作有重要提示意义。对于SIHIWES的临床诊断,当患者存在特殊面容、运动功能降低、肌骨发育异常、癫痫病史、性腺发育异常等,应高度考虑遗传性疾病,CHD4突变相关疾病不容忽视。对于SIHIWES患者的管理中,检测肌肉容积、骨骼发育并及时干预十分必要,这有利于进一步提高患者的生活质量,避免严重并发症的发生。同时,考虑到心律失常是癫痫严重合并症之一, 且CHD4是心脏、大脑共表达基因,对于确诊或疑诊SIHIWES的患者,除头颅MRI、脑电图等神经内科常规检查,心电图及心脏彩超也应纳入常规筛查项目。
参考文献:
1. WEISS K, TERHAL P A, COHEN L, et al. De novo mutations in CHD4, an ATP-dependent chromatin remodeler gene, cause an intellectual disability syndrome with distinctive dysmorphisms[J]. Am J Hum Genet, 2016, 99(4): 934-941.
2. SIFRIM A, HITZ M P, WILSDON A, et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing[J]. Nat Genet, 2016, 48(9):1060-1065.
3. WEISS K, LAZAR H P, KUROLAP A, et al. The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis[J]. Genet Med, 2020, 22(2): 389-397.
4. POLO S E, KAIDI A, BASKCOMB L, et al. Regulation of DNA‐damage responses and cell‐cycle progression by the chromatin remodelling factor CHD4[J]. EMBO J, 2010, 29(18): 3130-3139.
5. BASTA J, RAUCHMAN M. The nucleosome remodeling and deacetylase complex in development and disease[J]. Transl Res, 2015, 165(1): 36-47.
6. 周新龙,王清明,刘彦慧,等. 一例Sifrim-Hitz-Weiss综合征患儿的临床与CHD4基因变异分析[J]. 中华医学遗传学杂志, 2021, 38(1): 63-66.
7. LIU X R, YE T T, ZHANG W J, et al. CHD4 variants are associated with childhood idiopathic epilepsy with sinus arrhythmia[J]. CNS Neurosci Ther, 2021, 27(10): 1146-1156.
8. ROBBE Z L, SHI W, WASSON L K, et al. CHD4 is recruited by GATA4 and NKX2-5 to repress noncardiac gene programs in the developing heart[J]. Genes Dev, 2022, 36(7-8): 468-482.
9. WILCZEWSKI C M, HEPPERLA A J, SHIMBO T, et al. CHD4 and the NuRD complex directly control cardiac sarcomere formation[J]. Proc Natl Acad Sci U S A, 2018, 115(26): 6727-6732.
【引用格式】吴雨晨,钱方媛,张诗瑶,等. 成年型Sifrim-Hitz-Weiss综合征1例[J]. 中国神经精神疾病杂志,2025,51(1):32-37.
【Cite this article】WU Y C,QIAN F Y,ZHANG S Y, et al.A case of adult-type Sifrim-Hitz-Weiss syndrome[J]. Chin J Nervous Mental Dis,2025,51(1):32-37.
DOI:10.3969/j.issn.1002-0152.2025.01.008

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





该综合征表现为全方面的发育迟缓,主要临床表现有轻中度智力障碍、身材矮小、特殊面容等@
5
1例Sifrim-Hitz-Weiss综合征(Sifrim-Hitz-Weiss syndrome, SIHIWES)患者。患者为35岁男性,出生后有发育迟缓、肌肉萎缩、骨骼畸形、隐睾等症状,有宽前额、方形脸、低位耳、杯状耳、眼睑下垂等特殊面容。
1
#Sifrim-Hitz-Weiss综合征# #CHD4神经发育障碍#
8