
合并嗜酸性粒细胞增多,出现Purtscher样视网膜病变的非典型溶血性尿毒症一例
2025-03-21 协和医学杂志 协和医学杂志 发表于陕西省

本文报道1例合并嗜酸性粒细胞增多症aHUS患者的诊疗历程,并结合文献复习,以期提高临床对该病的认知。
非典型溶血性尿毒症(aHUS)是一种由补体旁路调节蛋白表达异常所致的危重性疾病,可引发微血管病性溶血性贫血、血小板减少症和急性肾损伤。除上述较为常见的临床三联征外,aHUS导致的缺血性器官损伤还可累及神经、心脏、肝脏、胰腺、消化道和眼部等多个部位,病变范围较广[1]。据国外调查显示,aHUS年发病率约为(0.23~1.9)/100万,占溶血尿毒综合征(HUS)病例的5%~10%,临床十分罕见[2-4]。国内方面,基于中国医院质量监测系统数据的分析显示,2018年中国aHUS发病率为0.038/100万,至2022年已增长为 2.996/100万[5]。本文报道1例合并嗜酸性粒细胞增多症aHUS患者的诊疗历程,并结合文献复习,以期提高临床对该病的认知。
病例资料
患者45岁女性,因“皮疹、发热、关节痛1个月余,少尿伴视物模糊3 d”于2023年12月3日就诊于北京协和医院急诊科。
2023年10月20日起,患者无明显诱因出现全身红色斑丘疹,伴瘙痒,否认口干、眼干、口腔溃疡、关节痛、脱发等,遂就诊于当地医院。查血常规:白细胞 5.89×109/L,嗜酸性粒细胞0.44×109/L(↑),血红蛋白 145 g/L,血小板 305×109/L。10月23日起出现发热(最高体温40 ℃),以午后发热为主,伴畏寒、寒战。先后予卢帕他定、依巴斯汀、头孢丙烯、罗红霉素、米诺环素治疗,上述症状无好转。
2023年11月1日起出现多处关节疼痛,累及双膝关节、双肩关节、双肘关节、双手近端指间关节,自服布洛芬后疼痛可减轻,但发热、皮疹无明显好转。此后,患者于多家医院就诊,查免疫指标、肿瘤指标、新型冠状病毒、甲型流感病毒、巨细胞病毒等均无殊。
2023年11月25日复查白细胞15.8×109/L(↑),嗜酸性粒细胞百分比2.3×109/L(↑),血红蛋白123 g/L,血小板228×109/L。
2023年11月29日至北京协和医院风湿免疫科就诊,查抗核抗体谱、系统性血管炎相关自身抗体谱均阴性。
2023年12月1日起,患者出现少尿(每天尿量少于400 mL),并出现进行性水肿症状,自双下肢至颜面部逐渐加重,伴视力显著下降,否认视野缺失、减小。11月30日、12月2日、12月3日于北京协和医院急诊科就诊,期间予对症支持治疗,相关检测结果见表1。
表1 2023年患者病情进展后相关检测指标变化
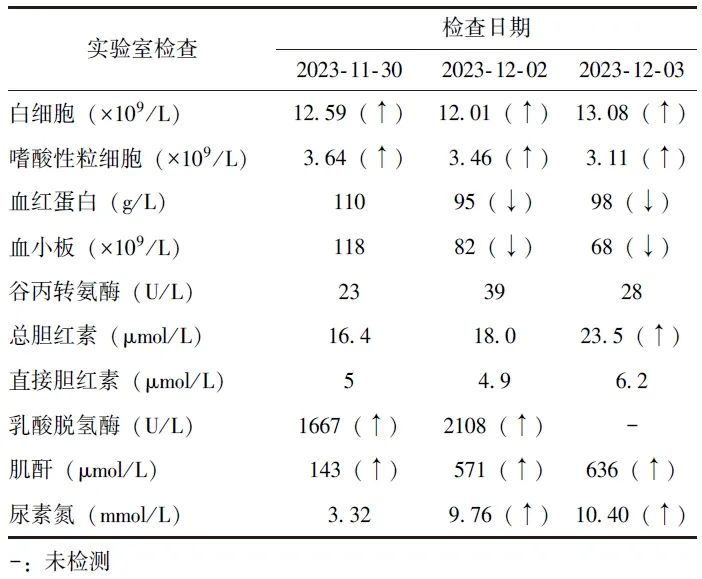
考虑“嗜酸性粒细胞升高、急性肾功能衰竭、血小板减少、不明原因发热待查”。因患者视力下降显著,眼科会诊发现双眼视盘周围大片浓密白色病灶,周边可见血管异常改变(图1),考虑“Purtscher样视网膜病变可能性大”,符合血栓性微血管病(TMA)病变的特征。为筛查TMA病因,完善血涂片并送ADAMTS13活性及其抑制物检测。
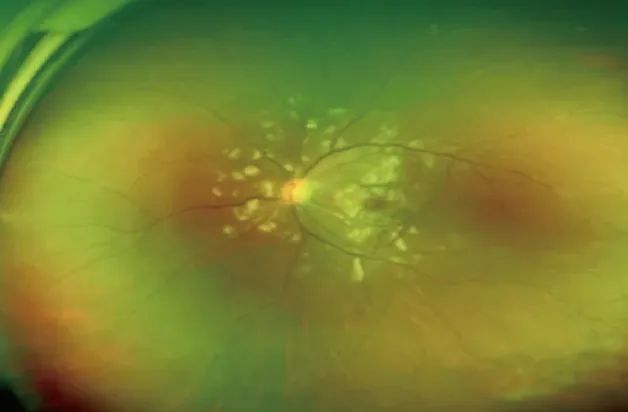
图1 患者眼底检查黄斑区呈半透明状,表面有血管,靠近视网膜中层,考虑Purtscher斑(2023-12-02)
2023年12月3日收住北京协和医院急诊重症监护室住院治疗。起病以来,患者精神、食欲差,大便干结,小便如上述,体质量增加5 kg。
既往史:2个月前颈部疼痛、僵硬,自认为颈椎病,予以热敷贴10余次(否认含有药物成份),疼痛得到缓解。否认近视。有青霉素皮试过敏史。
婚育史:适龄结婚,育有一女(20余年前剖宫产手术史)。
家族史:母亲健在,患有乳腺癌,父亲死于淋巴瘤。
入室查体:体温36.5 ℃,心率93次/min,呼吸频率16次/min,血压135/85 mm Hg(1 mm Hg=0.133 kPa),指氧饱和度100%(不吸氧状态)。格拉斯哥昏迷量表(GCS)评分:E4V5M6。全身皮肤弥漫色素沉着,表面呈细鳞屑状,巩膜轻度黄染;右侧腋窝可及质硬、肿大淋巴结(直径约3 cm),活动度尚可,有压痛;眼睑、四肢重度水肿;双眼视物模糊伴重影,视野正常;心、肺、腹部无明显阳性体征。
完善相关检查:血常规示白细胞12.26×109/L(↑),血红蛋白 83 g/L(↓),嗜酸性粒细胞2.81×109/L(↑),血小板58×109/L(↓);血浆游离血红蛋白254 mg/L(↑);直接抗球蛋白(coombs)试验阴性;谷丙转氨酶21 U/L,血清白蛋白29 g/L(↓),总胆红素23.2 μmol/L(↑),直接胆红素7.9 μmol/L;乳酸脱氢酶1502 U/L(↑);血肌酐604 μmol/L(↑),尿素氮10.2 mmol/L(↑),尿常规(导管尿)示尿蛋白≥3.0 g/L(↑),红细胞80/μL(↑);24 h尿蛋白0.58 g(尿量405 mL);动脉血气:pH 7.30(↓),血二氧化碳分压33 mm Hg(↓),氧分压131 mm Hg(↑),碳酸氢根15.7 mmol/L(↓),乳酸0.8 mmol/L;脑利尿钠肽 223 ng/L(↑),N末端脑利尿钠肽前体 3158 ng/L(↑);肌酸激酶54 U/L,肌酸激酶MB质量0.4 μg/L,肌红蛋白83 μg/L,高敏心肌肌钙蛋白Ⅰ<2.5 ng/L;电解质水平及凝血功能示除D-二聚体升高外,余指标均大致正常;炎症指标:超敏C反应蛋白15.89 mg/L(↑),降钙素原1.6 μg/L(↑),铁蛋白5897 μg/L(↑),白细胞介素-6/-8/-10均大致正常;总免疫球蛋白351 KU/L(↑)。外周血细胞涂片:可见较多红细胞碎片。血浆ADAMTS13活性100%,其抑制物为阴性。
患者血红蛋白水平进行性降低,游离血红蛋白、间接胆红素升高,有明确溶血证据,但coombs试验阴性,提示存在微血管病性溶血性贫血(MAHA)。结合患者合并血小板减少,外周血可见破碎红细胞,考虑存在TMA。由于血浆ADAMTS13活性正常,可排除血小板减少性紫癜(TTP)。为进一步筛查TMA病因,予完善肿瘤、免疫、感染相关检测(包括便培养、痰培养、外周血培养),结果均未见明显异常。超声示双侧腋窝多发淋巴结肿大、左侧颈部及锁骨上窝多发淋巴结皮质增厚、双侧腹股沟区多发淋巴结皮质增厚(因患者拒绝完善淋巴结活检未行病理检查)。
骨髓涂片:骨髓涂片可见粒系中性分叶核粒细胞比例(占比43.5%),嗜酸性粒细胞比例(占比26%)均增高,红细胞大小不一,大红细胞及嗜多色红细胞多见,并可见较多红细胞碎片及少许微小球形红细胞(图2)。

图2 患者骨髓涂片(2023-12-04)
A.可见粒系中性分叶核粒细胞比例、嗜酸性粒细胞比例均增高(×50);B.可见红细胞大小不一,大红细胞及嗜多色红细胞多见(×1000);C.可见较多红细胞碎片及少许微小球形红细胞(×1000)
骨髓病理:造血组织中粒红系比例增高,可见大量嗜酸性粒细胞。免疫组化:CD3(散在+),CD20(散在+),CD15(部分+),CD38(散在+),CD99(散在+),CD34(-),CD61(巨核细胞+),CD71(部分+),Ki-67(指数90%),MPO(部分+)。
由于患者感染、药物、代谢等因素介导的继发性TMA证据及其他全身性疾病证据不充分,考虑a-HUS可能性大。进一步查补体Ⅰ因子为196.72 μg/L(正常范围:12.26~333.02 μg/L)、H因子为201.84 μg/L(正常范围:246.60~417.69 μg/L),抗H因子抗体阴性。
综上,患者中年女性,急性起病,临床表现以肾功能急性恶化、伴视力进行性下降为主,血液学检测示贫血、血小板减少,间接胆红素、游离血红蛋白、乳酸脱氢酶升高,血涂片见大量破碎红细胞,眼底检查可见Purtscher样视网膜病变,提示TMA诊断明确。ADAMTS13活性正常而其抑制物为阴性,可排除TTP。
患者起病时无腹泻,便常规及便培养(包括志贺菌素等)均阴性,可排除产志贺毒素大肠杆菌溶血尿毒综合征(STEC-HUS);最后完善肿瘤、免疫疾病及其他感染相关指标筛查均无明确支持证据,因此最终诊断为aHUS。
遂完善sC5b-9定量:>2130 μg/L(正常范围:75~219 μg/L);血清总补体CH50 26.8 U/mL(正常范围:23~46 U/mL),以评估病情活动度。由于原发性aHUS常合并H因子基因突变,如存在高危突变或需终身治疗,遂送检全外显子检测,结果回报未见基因突变。
治疗:除常规支持治疗外,TMA-aHUS治疗方面,2023年12月3日予单膜血浆置换1次,2023年12月15日、2023年12月25日、2024年1月6日分别予依库珠单抗900 mg静脉输注,同期予头孢他啶(2 g,每12小时1次)静脉输注预防感染。由于患者病程中嗜酸性粒细胞增多持续时间小于4周,虽起病时伴皮疹,但皮疹出现在嗜酸性粒细胞增高前,且患者无高嗜酸性粒细胞增多症相关家族史,CD3-CD4+T细胞占淋巴细胞总数的0.24%,且无其他明确的嗜酸性粒细胞累及脏器的证据,因此考虑高嗜酸性粒细胞综合征证据不足,针对该患者嗜酸性粒细胞增多症的治疗主要为病因治疗,采用糖皮质激素疗法,方法如下:甲泼尼龙40 mg/次,每12小时给药1次(2023年12月4日起)→甲泼尼龙40 mg,每天1次(2023年12月12日起)→甲泼尼龙20 mg,每天1次(2023年12月21日起)→泼尼松20 mg,每天1次(2023年12月28日起)→泼尼松15 mg,每天1次(2024年1月7日起)→泼尼松10 mg,每天1次(1月13日起),此后计划每周减半片泼尼松至停药。
2024年1月1日,患者住院治疗期间出现发作性头部及躯干抖动,无意识丧失及其他神经系统体征变化,头颅CT、MRI及脑电图基本正常,神经内科会诊考虑为TMA相关肌震挛可能。加用氯硝西泮,2024年1月3日后该症状未再发作。患者住院期间先后出现肺部感染、胆囊炎、巨细胞病毒感染及甲流感染,予以对症治疗后均恢复正常。患者住院期间重要脏器功能指标变化情况见表2。
表2 患者住院期间重要脏器功能指标变化情况
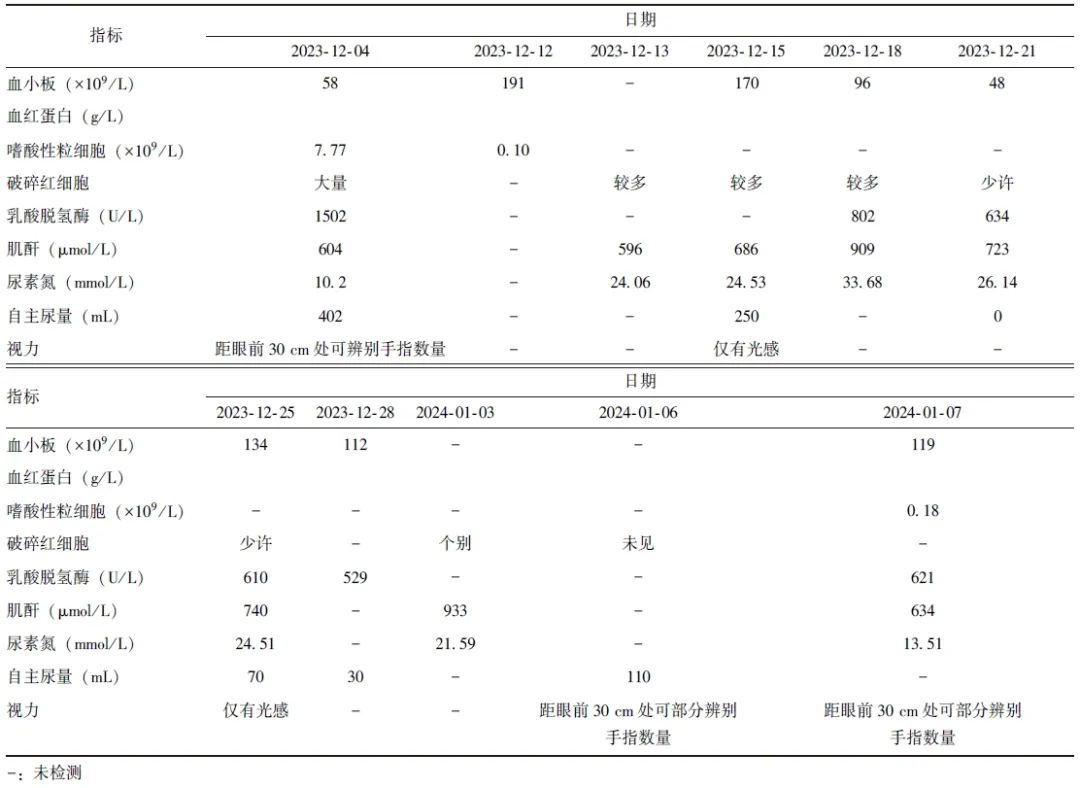
2024年1月11日,患者血小板水平逐渐恢复正常,血涂片未见破碎红细胞,规律血液透析情况下肾功能未出现恶化,且无其他新发脏器受累表现,考虑aHUS控制尚可,且嗜酸性粒细胞恢复正常、皮疹消失,提示病情趋于稳定,予以出院。出院后继续依库珠单抗治疗,嘱患者尽快完成脑膜炎双球菌疫苗接种。停用头孢他啶,改为头孢呋辛(0.25 g,每天2次)预防感染。2024年5月13日随访,患者已应用依库珠单抗11个疗程,肾脏仍需规律血液透析,监测肌酐水平为150~350 μmol/L,尿量约为400 mL/d。自诉视力较出院时无明显好转。外院查血红蛋白95 g/L,血小板恢复正常,生活可自理。
讨论
HUS是指临床表现为微血管病性溶血性贫血、血小板减少和急性肾损伤的一组临床综合征[6]。根据2016年国际HUS分类[7],HUS可分为感染(肺炎链球菌、流感病毒及产志贺毒素大肠杆菌等)相关HUS、钴胺素C 缺乏相关 HUS、aHUS及继发性HUS[8-9]。其中,aHUS又称为补体系统介导的TMA,是遗传性或获得性补体调节缺陷导致补体旁路过度活化所引发的以肾脏受累最为常见的多系统损害临床综合征[2,10],约50%患者最终进展为终末期肾病(ESKD)[1]。在aHUS的发病机制中,补体H因子、补体I因子和膜辅助蛋白等替代补体通路调节因子发挥重要作用,已有报道发现aHUS患者中编码这些补体调节蛋白的基因存在异常[11]。
aHUS为罕见病,已被纳入我国卫生健康委2018年公布的《第一批罕见病目录》[6]。由于aHUS发病机制主要由于补体蛋白基因突变或机体存在补体蛋白抗体,经触发事件(例如感染、妊娠、药物、肿瘤、器官移植等)引起补体替代途径不可抑制的持续激活,从而导致膜攻击复合物形成,肾脏内皮损伤、凝血级联活化和肾小球动脉微血栓形成[12],因此患者肾脏受累较为突出。本例患者在急诊科就诊的3天期间,在无明确肾前性或肾后性因素的影响下,肾功能急剧恶化,肌酐、尿素氮迅速升高至需血液净化的水平,自主尿量由少尿亦快速进展至无尿状态,肾脏病变尤为显著,难以用一般疾病进行解释。
除肾脏受累外,该病还可广泛累及神经、心脏、肝脏、胰腺、消化道和眼等多个脏器,预后不佳[1]。其中,最常见的肾外受累部位是神经系统,常以偏瘫、视力丧失、头痛和幻觉为主要症状[13]。少数文献报道了aHUS眼部受累症状,多表现为视力下降、眼部疼痛、复视和视力模糊等[14-15]。本例患者因急进性视力损害筛查发现典型Purtscher样视网膜病变而为明确诊断提供了重要线索。
1910年Otmar Purtscher首次描述了Purtscher视网膜病变[16],其是指胸腹部严重挤压伤或粉碎性骨折后发生的一种特殊的视网膜病理改变[17],眼底表现包括棉绒斑、伴或不伴有中低数量视网膜出血、Purtscher斑、黄斑水肿。50%病例可出现特征性Purtscher斑,多分布于黄斑区和视盘鼻侧,其与毛细血管前小动脉的阻塞有关[18]。
Purtscher样视网膜病变则是在非创伤的情况下出现的类似于Purtscher视网膜病变的眼底表现,常与系统性红斑狼疮、急性胰腺炎、眼眶和鼻道内及周围注射类固醇等引起广泛微血栓形成,继而导致毛细血管前小动脉阻塞相关,其为一种非常罕见的视网膜疾病,年发病率为0.24/10万。2017年文献报道了1例因“急性双侧视力模糊和头痛、头晕和晕厥病史3 d”而就诊的25岁女性患者,此为首例报道的与aHUS 相关的Purtscher 样视网膜病变病例[19]。其预后方与累及部位有关。本例患者累及黄斑区,视力预后欠佳。
此外,本例患者特别之处在于其合并嗜酸性粒细胞增多症。起病初期出现全身红色斑丘疹。皮疹可为嗜酸性粒细胞增多症的首发表现。一项多中心回顾性研究报告显示,37%的嗜酸性细胞增多症患者以皮疹为首发和最常见症状,临床表现为湿疹、泛发性皮肤增厚、皮肤划痕征等[20]。嗜酸性粒细胞增多症与 TMA 的关联性尚不明确。部分研究者认为,嗜酸性粒细胞脱颗粒可在肾微血管系统中引起内皮损伤,从而诱发TMA[21]。既往文献中已有嗜酸性粒细胞增多症和TMA同时存在的病例报道,如Fukui等[22]曾诊治了1例合并嗜酸性粒细胞增多症的TMA病例。个别aHUS患儿病程中可见嗜酸性粒细胞增多[23]。
综上,嗜酸性粒细胞增多症与aHUS可能存在一定关联或可能具有共同的遗传基础,当患者暴露于某些环境因素时,此两种疾病才会显现,但目前相关证据尚不充分,二者的关系尚需更多证据阐明[11,24]。针对嗜酸性粒细胞增多症,该患者采用激素疗法治疗,病程中监测患者的嗜酸性粒细胞水平逐渐恢复正常,全身皮疹逐渐消退且再无新发皮疹。
《中国儿童非典型溶血尿毒综合征诊治专家共识(2023版)》推荐[1],一旦怀疑或确诊aHUS,除常规对症支持治疗,如输注红细胞、补液、透析等,还应在24~48 h内启用依库珠单抗单抗。对于无法获得该药的患者,建议采用血浆置换治疗[7,12,25]。2009年Nürnberger 等[26]首次报道使用人源化 C5单克隆抗体依库珠单抗成功治疗1例aHUS患者的案例。临床研究发现[27],依库珠单抗治疗aHUS起效迅速,对于血液学处于活动期的急性患者,治疗第2周即可观察到血小板计数显著改善,对于起病时需肾脏替代的急性肾损伤患者,75%在治疗7.7 d(中位时间)内脱离透析治疗,后期进展为ESKD的风险显著降低(约为11.8%)。一项回顾性研究纳入了72例接受依库珠单抗治疗的aHUS患者,中位随访2.1年时整体病死率为1.4%,几乎所有患者均达到了蛋白尿缓解,透析依赖患者比例由起病时的38%降至10%[28]。本例患者亦采用了依库珠单抗治疗,截至末次随访已治疗11个疗程,监测每日尿量约为400 mL/d,但仍无法脱离血液透析支持,血肌酐、尿素氮水平在透析支持下趋于稳定。
综上,本文报道了1例累及肾脏、眼睛、血液系统及神经系统且合并嗜酸性粒细胞增多症的罕见aHUS病例。aHUS作为非特异性疾病,在筛查过程中需充分排除感染、肿瘤、免疫、药物、代谢等继发因素,并结合补体水平监测及基因检测结果综合考虑,从而明确诊断。该病受累脏器多,临床表现复杂,提示临床在接诊多系统受累的患者时,除对常见全身性疾病的考量外,还应警惕罕见疾病的可能性。
作者贡献
梅琦敏负责数据整理及文章撰写;戴佳原负责数据整理及文章修改;刘业成、沈敏、朱华栋负责论文审核。
参考文献
[1]中国罕见病联盟儿童非典型溶血尿毒综合征专业委员会, 国家儿童医学中心(首都医科大学附属北京儿童医院), 《中华实用儿科临床杂志》编辑委员会. 中国儿童非典型溶血尿毒综合征诊治专家共识 (2023版)[J]. 中华实用儿科临床杂志, 2023, 38(6): 401-412.
[2]Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults[J]. Clin J Am Soc Nephrol, 2013, 8(4): 554-562.
[3]Wühl E, Van Stralen K J, Wanner C, et al. Renal replacement therapy for rare diseases affecting the kidney: an analysis of the ERA-EDTA Registry[J]. Nephrol Dial Transplant, 2014, 29(Suppl 4): iv1-iv8.
[4]Bayer G, Von Tokarski F, Thoreau B, et al. Etiology and outcomes of thrombotic microangiopathies[J]. Clin J Am Soc Nephrol, 2019, 14(4): 557-566.
[5]Zheng H, Chen L .Epidemiological and Clinical Characteristics of Atypical Hemolytic Uremic Syndrome in China[J].J Am Soc Nephrol, 2024.DOI:10.1681/ASN.2024m9jgqqwn.
[6]张抒扬. 中国第一批罕见病目录释义[M]. 北京: 人民卫生出版社, 2018: 364-366.
[7]Loirat C, Fakhouri F, Ariceta G, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children[J]. Pediatr Nephrol, 2016, 31(1): 15-39.
[8]Fakhouri F, Zuber J, Frémeaux-Bacchi V, et al. Haemolytic uraemic syndrome[J]. Lancet, 2017, 390(10095): 681-696.
[9]Le Clech A, Simon-Tillaux N, Provt F, et al. Atypical and secondary hemolytic uremic syndromes have a distinct presentation and no common genetic risk factors[J]. Kidney Int, 2019, 95(6): 1443-1452.
[10]Mizuno K, Dandoy C E, Teusink-Cross A, et al. Eculizu-mab precision-dosing algorithm for thrombotic microangiopa-thy in children and young adults undergoing HSCT[J]. Blood Adv, 2022, 6(5): 1454-1463.
[11]Cao M, Ferreiro T, Leite B N, et al. Two cases of atypical hemolytic uremic syndrome (aHUS) and eosinophilic granulomatosis with polyangiitis (EGPA): a possible relationship[J]. CEN Case Rep, 2017, 6(1): 91-97.
[12]李璐, 毛建华. 溶血尿毒综合征的诊断及治疗进展[J]. 中华实用儿科临床杂志, 2021, 36(17): 1285-1289.
[13]Timmermans S A M E G, Van Paassen P. The syndromes of thrombotic microangiopathy: a critical appraisal on complement dysregulation[J]. J Clin Med, 2021, 10(14): 3034.
[14]Tzoumas N, Hallam D, Harris C L, et al. Revisiting the role of factor H in age-related macular degeneration: Insights from complement-mediated renal disease and rare genetic variants[J]. Surv Ophthalmol, 2021, 66(2): 378-401.
[15]Yenerel M N. Atypical hemolytic uremic syndrome: differential diagnosis from TTP/HUS and management[J]. Turk J Haematol, 2014, 31(3): 216-225.
[16]Vezzola D, Allegrini D, Romano M R, et al. Optical coherence tomography angiography in Purtscher-like retinopathy associated with dermatomyositis: a case report[J]. J Med Case Rep, 2019, 13(1): 206.
[17]戴荣平, 董方田, 郑霖, 等. Purtscher样视网膜病变[J]. 中华眼科杂志, 2007, 43(5): 447-450.
[18]Stroh I, Nguyen A, Channa R. Purtscher-like retinopathy in hemolytic uremic syndrome[J]. JAMA Ophthalmol, 2019, 137(1): e183911.
[19]Ustaoğlu M, Önder F, Solmaz N, et al. Purtscher-like retinopathy associated with atypical hemolytic uremic syndrome[J]. Turk J Ophthalmol, 2017, 47(6): 348-350.
[20]Williams K W, Ware J, Abiodun A, et al. Hypereosino-philia in children and adults: a retrospective comparison[J]. J Allergy Clin Immunol Pract, 2016, 4(5): 941-947.e1.
[21]Yuste C, Quiroga B, Verde E, et al. The non-casual relation between eosinophilia and thrombotic microangiopathy[J]. Transfus Apher Sci, 2012, 47(3): 365-367.
[22]Fukui S, Iwamoto N, Tsuji S, et al. Eosinophilic granulomatosis with polyangiitis with thrombotic microangiopathy: Is simultaneous systemic lupus erythematosus associated with clinical manifestations?: A case report and review of the literature[J]. Medicine (Baltimore), 2015, 94(45): e1943.
[23]Ito N, Hataya H, Saida K, et al. Efficacy and safety of eculizumab in childhood atypical hemolytic uremic syndrome in Japan[J]. Clin Exp Nephrol, 2016, 20(2): 265-272.
[24]Cataland S R, Wu H M. How I treat: the clinical differentiation and initial treatment of adult patients with atypical hemolytic uremic syndrome[J]. Blood, 2014, 123(16): 2478-2484.
[25]De Souza R M, Correa B H M, Melo P H M, et al. The treatment of atypical hemolytic uremic syndrome with eculizumab in pediatric patients: a systematic review[J]. Pediatr Nephrol, 2023, 38(1): 61-75.
[26]Nürnberger J, Philipp T, Witzke O, et al. Eculizumab for atypical hemolytic-uremic syndrome[J]. N Engl J Med, 2009, 360(5): 542-544.
[27]Legendre C M, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome[J]. N Engl J Med, 2013, 368(23): 2169-2181.
[28]Muff-Luett M, Sanderson K R, Engen R M, et al. Eculizumab exposure in children and young adults: indications, practice patterns, and outcomes-a Pediatric Nephrology Research Consortium study[J]. Pediatr Nephrol, 2021, 36(8): 2349-2360.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








#嗜酸性粒细胞增多症# #非典型溶血性尿毒症#
7