
Nature Medicine:杜氏肌营养不良症的AAV基因疗法
2024-10-17 MedSci原创 MedSci原创 发表于上海

尽管delandistrogene moxeparvovec在主要终点NSAA评分变化上未能显示出统计学显著优势,但在微抗肌萎缩蛋白表达和其他次要终点上显示出一定的积极趋势。
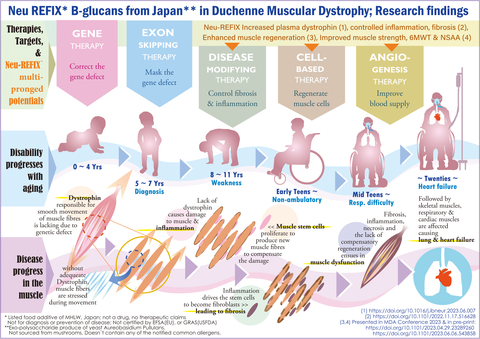
杜氏肌营养不良症(Duchenne Muscular Dystrophy, DMD)是一种罕见的X连锁神经肌肉疾病,由DMD基因中的致病变异引起,导致功能性抗肌萎缩蛋白缺失。这种缺失从出生开始,逐渐导致运动功能受损、丧失行走能力以及威胁生命的呼吸和心脏并发症。Delandistrogene moxeparvovec是一种基于腺相关病毒rh74载体的基因疗法,旨在解决DMD患者中缺乏的功能性抗肌萎缩蛋白问题。

EMBARK研究是一项III期临床试验,旨在评估delandistrogene moxeparvovec在DMD患者中的疗效和安全性。该研究纳入了年龄在4至8岁以下能够行走的男性DMD患者,根据年龄组和北星步行评估(North Star Ambulatory Assessment, NSAA)评分进行随机分层,接受单次静脉注射delandistrogene moxeparvovec(1.33 × 10^14 基因组每千克;n=63)或安慰剂(n=62)。主要终点是第52周时NSAA评分较基线的变化。
研究发现,第52周时,NSAA评分变化未达到统计学显著差异。delandistrogene moxeparvovec组和安慰剂组的最小二乘均值变化分别为2.57分和1.92分,组间差异为0.65分(95%置信区间,-0.45至1.74;P=0.2441)。
第12周时,微抗肌萎缩蛋白表达量:治疗组为34.29%,安慰剂组为0.00%。第52周时,各次要终点的组间差异(95%置信区间)包括:起立时间(-0.64秒,-1.06至-0.23秒)、10米步行/跑步时间(-0.42秒,-0.71至-0.13秒)、95百分位步速(0.10米/秒,0.00至0.19米/秒)、100米步行/跑步时间(-3.29秒,-8.28至1.70秒)、上四阶楼梯时间(-0.36秒,-0.71至-0.01秒)、PROMIS移动性和上肢功能(0.05分,-0.08至0.19分;-0.04分,-0.24至0.17分)以及NSAA技能获得或改善的数量(0.19项,-0.67至1.06项)。
此次研究总共记录了674例delandistrogene moxeparvovec组的不良事件和514例安慰剂组的不良事件。没有死亡、停药或临床上显著的并发症报告。特别关注的不良事件包括丙氨酸氨基转移酶(ALT)和天冬氨酸氨基转移酶(AST)水平升高,但这些事件大多发生在输注后90天内,并且没有导致严重的后果。
由于主要终点未达到统计学显著性,且统计分析计划中没有对多重性进行校正的规定,因此结果以点估计值和组间差异的最小二乘均值变化及95%置信区间的形式报告。为了全面评估证据并解决多重假设检验的问题,还进行了预先指定的探索性疗效分析,使用了Wei-Lachin程序,将多个终点的信息综合成一个单一的全球统计检验,以评估总体治疗效果。
尽管delandistrogene moxeparvovec在主要终点NSAA评分变化上未能显示出统计学显著优势,但在微抗肌萎缩蛋白表达和其他次要终点上显示出一定的积极趋势。安全性方面,虽然观察到了一些肝功能指标的升高,但整体上是可管理的。未来的研究需要进一步探讨该基因疗法在DMD患者中的长期疗效和安全性。
参考文献:
Jerry R, Mendell,Francesco, Muntoni,Craig M, McDonald et al. AAV gene therapy for Duchenne muscular dystrophy: the EMBARK phase 3 randomized trial.[J] .Nat Med, 2024, 0: 0.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言











#杜氏肌营养不良症#
75