
“绘”解读真报告 | SMARCB1基因变异检测,辅助非典型畸胎样/横纹肌样瘤临床诊断和遗传风险评估
6小时前 苏州绘真医学 苏州绘真医学 发表于陕西省

本文介绍非典型畸胎样/横纹肌样瘤(AT/RT),通过1例4岁女童病例,阐述其诊断过程。文中提及基因检测作用,解释WHO分类下的诊断标准、基因胚系有害变异与肿瘤遗传风险,以及相关检测推荐。
非典型畸胎样/横纹肌样瘤(atypical teratoid/rhabdoid tumor,AT/RT)是一种罕见的高度恶性的中枢神经系统胚胎性肿瘤,约占儿童CNS肿瘤的1%-2%。90%以上的AT/RT发生在3岁以下儿童,成人患者只有少数病例报告[1]。由于临床表型无特异性,影像学表现多样性,初诊困难,需依靠病理学诊断。基因检测也有助于AT/RT的确诊,其特征为SMARCB1和SMARCA4基因突变[2]。
案例介绍
近日,1例4岁女童患者由于头痛、恶心等症状,入院治疗,临床诊断提示为颅内占位。免疫组化检测为INI-1(SMARCB1基因编码)蛋白表达缺失,病理诊断提示符合非典型畸胎样/横纹肌样瘤(AT/RT),WHO 4级,建议进一步做相关分子遗传学检测。

图1 病理报告要求进一步分子遗传学检测,辅助诊断
该患者选择送检我司实体瘤1299基因检测,该项目包括了SMARCB1基因和SMARCA4基因的全部编码区。样本类型为手术肿瘤组织+对照血。样本和测序质控均合格,其中肿瘤(异形)细胞占比为80%,平均测序深度为3365.24X。检测结果提示,该患者携带SMARCB1 p.R201*无义突变,变异丰度为91.93%。另外,还检出ARID1A p.D1850Gfs*4移码突变和DLG2 p.R765*无义突变。根据WHO分类,进一步辅助临床确诊该患者为非典型畸胎样/横纹肌样瘤。由于该SMARCB1基因变异为体系变异,提示肿瘤遗传风险低。
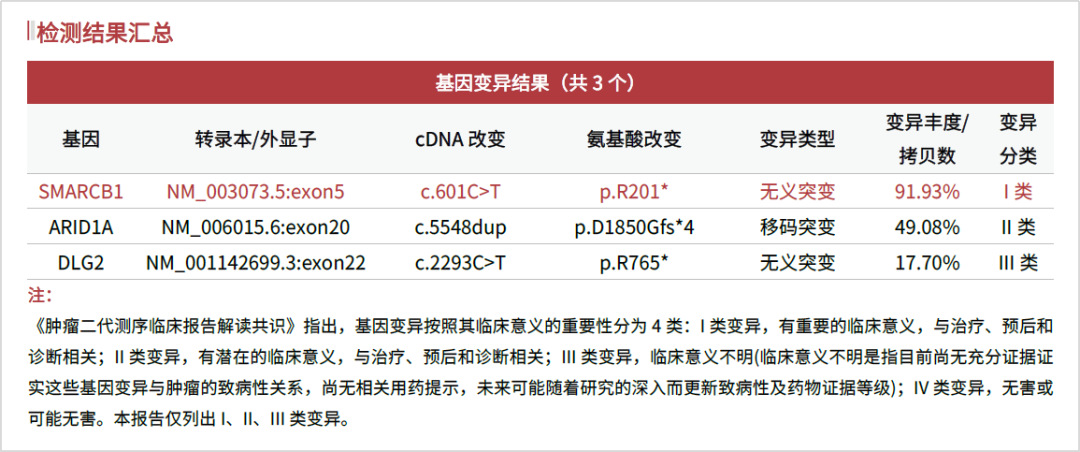
图2 绘真医学实体瘤1299基因检测项目检出SMARCB1双等位基因失活突变
一、WHO分类指出,SMARCB1或SMARCA4突变是AT/RT的理想诊断标准
《第5版WHO中枢神经系统肿瘤分类》指出,非典型畸胎样/横纹肌样瘤(AT/RT)是由低分化细胞和数量不等的横纹肌样细胞组成的高级别恶性肿瘤,具有沿神经上皮、上皮和间充质线分化的潜能。在遗传学上,常见SMARCB1(>95%)或SMARCA4(<5%)双等位基因失活突变。
AT/RT的基本诊断标准为:多种免疫表型的中枢神经系统胚胎性肿瘤,同时肿瘤细胞核SMARCB1或SMARCA4蛋白表达缺失,或者具有符合AT/RT的DNA甲基化谱。理想标准为:横纹肌细胞,且SMARCB1或SMARCA4基因突变[3]。

图3 WHO分类推荐SMARCB1或SMARCA4基因突变辅助AT/RT诊断
德国明斯特大学附属医院Martin Hasselblatt等人采用一种高分辨方法——分子倒置探针单核苷酸多态性(MIP SNP)分析16例SMARCB1蛋白表达缺失的AT/RT患者SMRACB1基因拷贝数变异,其中15例(94%)检出SMARCB1基因缺失或拷贝数平衡杂合性缺失变异(copy number neutral LOH)。同时采用FISH、MLPA、PCR方法检测SMARCB1基因拷贝数变异、大片段重排变异和点突变。4例携带拷贝数平衡杂合性缺失变异的患者,3例检出SMARCB1基因纯合突变,1例检出SMARCB1 5号外显子纯合缺失。剩余1例MIP SNP检测阴性的患者,MLPA方法检出SMARCB1 5-7号显子子杂合缺失变异[4]。
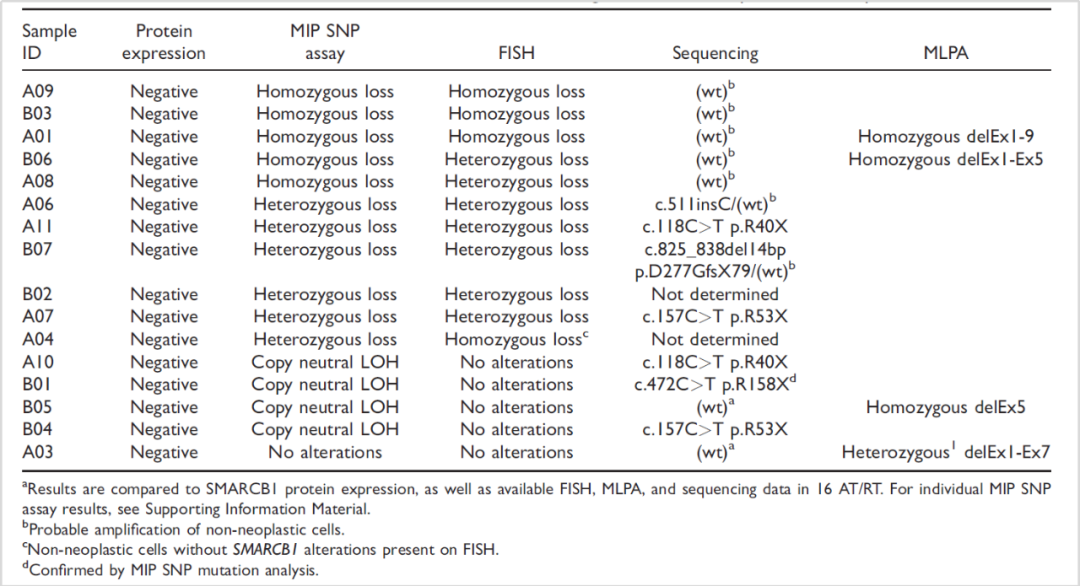
图4 16例AT/RT患者MIP SNP等方法检测SMARCB1基因变异结果
除了SMARCB1基因突变外,WHO分类指出,还有不足5%的AT/RT患者,组织学形态符合AT/RT,但SMARCB1蛋白表达,携带SMARCA4双等位基因失活突变和SMARCA4蛋白表达缺失。这类患者往往发病年龄很小,预后差。
德国汉堡大学医学中心汇总了14例甲基化检测分类为SMARCA4异常的AT/RT患者的SMARCA4基因变异和蛋白表达信息。免疫组化(IHC)检测提示,12例SMARCA4蛋白表达缺失, 11例检出SMARCA4基因变异,包括5例无义突变,3例移码突变,1例杂合性缺失,1例SMARCA4基因断裂变异,1例内含子区剪接突变。2例SMARCA4蛋白表达的AT/RT患者,均检测SMARCA4基因错义突变[5]。
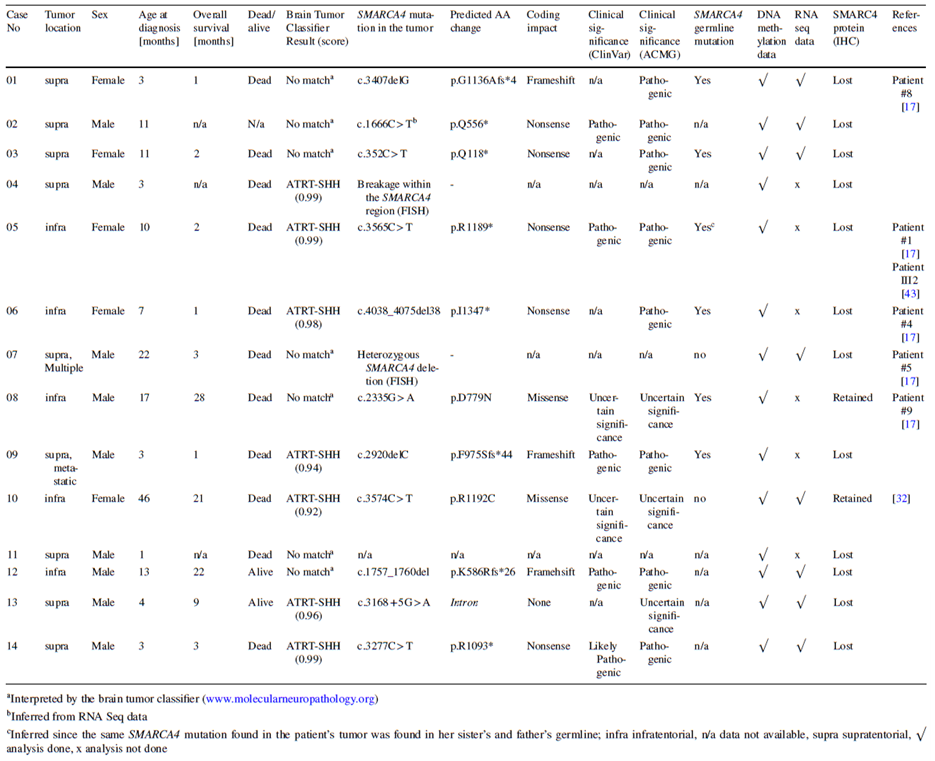
图5 14例AT/RT患者的SMARCA4基因变异和蛋白表达情况
二、SMARCB1或SMARCA4基因胚系有害变异,提示肿瘤遗传风险
WHO分类指出,家族性AT/RT患者通常由横纹肌样瘤易感综合征1(rhabdoid tumour predisposition syndrome 1,RTPS1)或横纹肌样瘤易感综合征1(rhabdoid tumour predisposition syndrome 2,RTPS2)导致,对应的基因分别为SMARCB1和SMARCA4。SMARCB1缺失AT/RT患者,胚系突变的发生率为26%至41%,SMARCA4缺失的AT/RT患者,胚系突变率更高[3]。
RTPS是一种具有高度发展为恶性横纹肌样瘤和/或非典型畸胎样/横纹肌样瘤风险的疾病。组织学上,RTPS与散发性患者表现相似。其主要诊断标准为MRT或AT/RT患者存在SMARCB1或SMARCA4基因胚系突变。次要诊断标准为多发性MRT(恶性横纹肌瘤)或AT/RT,同胞或其他亲属患有MRT或AT/RT[6]。
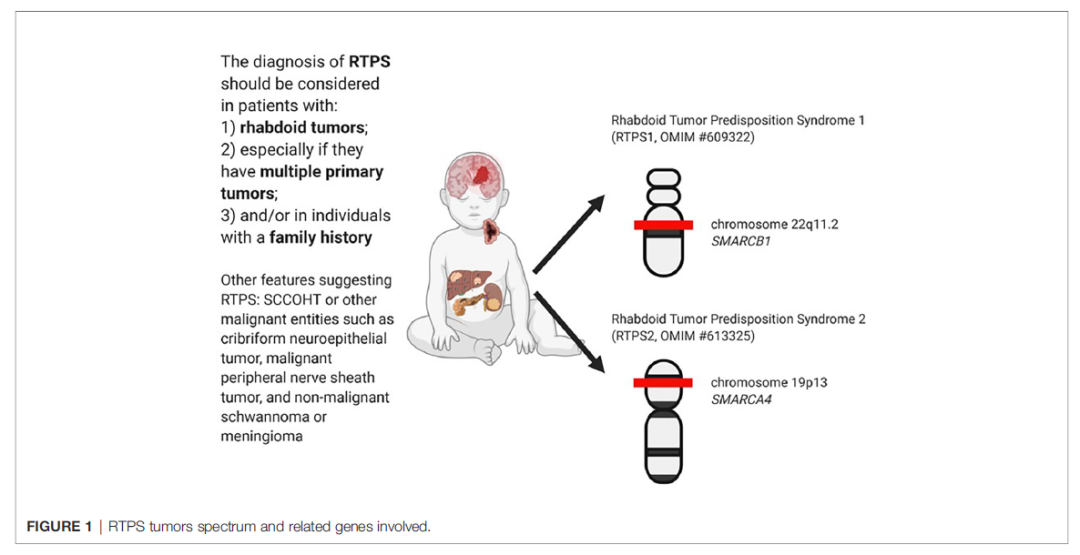
图6 AT/RT遗传相关的横纹肌样瘤易感综合征(RTPS)及基因变异
SMARCB1基因位于染色体22q11.2区域,包括9个外显子区。变异类型多样,包括整条染色体异常、22q11.2区域杂合性缺失、功能性缺失突变,例如无义突变、移码突变、剪接突变和错义突变。AR/RT最常见的是22号染色体单体。SMARCA4基因位于染色体19p13区域,包括36个外显子区,也有多种变异类型。无义突变和基因内缺失最为常见,也有检出错义突变[7]。
为了实现在肿瘤还有可能完全切除的时就确诊,the SIOPE Host Genome工作组关于AT/RT遗传基因检测做如下推荐:①所有的AT/RT患者进行胚系基因变异检测,无论年龄。②对于携带SMARCB1或SMARCA4基因胚系有害变异的患者,需进行专家遗传咨询,无论年龄。③对于携带SMARCB1或SMARCA4基因胚系有害变异的患者的所有一级亲属,进行SMARCB1或SMARCA4基因检测和遗传咨询,无论年龄,包括了产前检测。④对于携带SMARCB1或SMARCA4基因胚系变异的患者和一级亲属,应尽可能早开始临床随访。⑤临床随访至少包括超声和临床检查,全身和中枢神经系统核磁共振检查(MRI)的间隔期范围变化较大,与所在机构的资源以及临床医生偏好相关[8]。

图7 工作组推荐AT/RT患者及其所有一级亲属进行SMARCB1和SMARCA4基因胚系变异检测
绘真医学实体瘤全外显子组基因检测和实体瘤1299基因检测项目,覆盖了SMARCB1和SMARCA4基因的全部编码区,以及外显子和内含子毗邻区域±20bp,可以检测点突变、小片段插入缺失以及拷贝数变异,能够辅助AT/RT患者的临床诊断和遗传风险评估。
参考文献:
[1]高文龙,苟攀,梁平.儿童中枢神经系统非典型畸胎样/横纹肌样瘤研究进展[J].中国神经精神疾病杂志, 2023, 49(11):682-688.
[2]王蒙,赵毅,赵培超,等. 儿童颅内非典型畸胎样/横纹肌样瘤的诊治及预后分析[J]. 中华实用儿科临床杂志,2021,36(10):748-752.
[3]WHO Classification of Tumours Editorial Board. World Health Organization Classification of Tumours of the Central Nervous System.5th ed. Lyon: International Agency for Research on Cancer; 2021.
[4]Hasselblatt, Martin et al. “High-resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors.” Genes, chromosomes & cancer vol. 52,2 (2013): 185-90. doi:10.1002/gcc.22018
[5]Holdhof, Dörthe et al. “Atypical teratoid/rhabdoid tumors (ATRTs) with SMARCA4 mutation are molecularly distinct from SMARCB1-deficient cases.” Acta neuropathologica vol. 141,2 (2021): 291-301. doi:10.1007/s00401-020-02250-7
[6]方园,陈莲,何乐健. 第5版WHO儿童肿瘤分类遗传性肿瘤综合征解读(一)[J]. 中华病理学杂志,2023,52(12):1197-1203.
[7]Del Baldo, Giada et al. “Rhabdoid Tumor Predisposition Syndrome: From Clinical Suspicion to General Management.” Frontiers in oncology vol. 11 586288. 22 Feb. 2021, doi:10.3389/fonc.2021.586288
[8]Frühwald, M C et al. “Current recommendations for clinical surveillance and genetic testing in rhabdoid tumor predisposition: a report from the SIOPE Host Genome Working Group.” Familial cancer vol. 20,4 (2021): 305-316. doi:10.1007/s10689-021-00229-1

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





前往app查看评论内容
0 0