
Nature Genetics:海鲜啤酒并不是造成痛风的主要原因!GWAS阐明痛风背后的真相
2024-11-14 梅斯学术 梅斯学术 发表于江苏省

青岛大学等多机构研究人员通过 260 万人 GWAS 研究痛风,介绍研究内容如痛风基因座等方面,确定相关基因位点等,跨种族分析增强结果可靠性,为痛风遗传学及治疗提供新策略。
痛风是一种慢性疾病,是由于尿酸水平高而引起的“关节炎”。人们往往认为痛风是生活方式或饮食引起的(例如吃海鲜、喝啤酒),因而被痛风困扰的人们通常会克制自己的饮食,减少海鲜、啤酒等食物或饮品的摄入。但真实情况果真如此吗?饮食习惯是造成痛风的主要原因吗?
近日,青岛大学师咏勇教授、李长贵教授联合阿拉巴马大学伯明翰分校、奥塔哥大学、23andMe公司等机构的研究人员,在Nature子刊Nature Genetics上发表了研究成果,从260万人的全基因组关联研究(GWAS)中提供了对痛风炎症成分的分子机制的见解,包括120,295名流行痛风患者。
该研究通过优先级方案确定了痛风炎症过程中的候选基因,提出迄今为止最大的痛风GWAS的研究结果,优先考虑先前未确定的痛风炎症过程候选途径,为后续研究提供理论基础。

在开始今天的研究解读之前,先带大家了解一下GWAS,可能不少读者对它“一知半解”——仅听过GWAS的大名,但不了解它“是什么”“怎么用”“如何发文”。
提到GWAS,就不得不提到孟德尔随机化。获取相关疾病的GWAS数据,是顺利进行孟德尔随机化的关键之一,进而能探讨不同疾病间的关联或确定疾病的致病基因。
GWAS的全称是Genome-wide association studies,即全基因组关联研究,是一种用于识别遗传区域(基因组)和性状/疾病之间关联的方法。通过检测各类生物基因组中数百或上千万的遗传变异,GWAS能够找出那些与特定表型或疾病具有显著关联的变异位点。

GWAS分析基本内容(图源:生信大碗)
接下来,我们用Nature子刊上的实际案例,来分析一下GWAS相关研究该如何开展。
一、研究内容与思路
1、痛风基因座、遗传风险预测及基因精细定位
这项研究参与者来自13个队列,对四个祖先群体以及跨祖先群元(TAMA)进行全基因组关联研究(GWAS)分析,每个分析都包括性别特异性分析。
TAMA共检测到295个全基因组显著性位点,其中20个位点有超过一个遗传独立信号,318个独立的全基因组显著性信号。
在单祖先群体GWAS分析中,在非洲人中检测到2个全基因组显著位点,在东亚人中检测到10个,在欧洲人中检测到276个,在拉丁人中检测到10个。性别分层GWAS分析发现男性特有的247个位点和女性特有的14个位点。
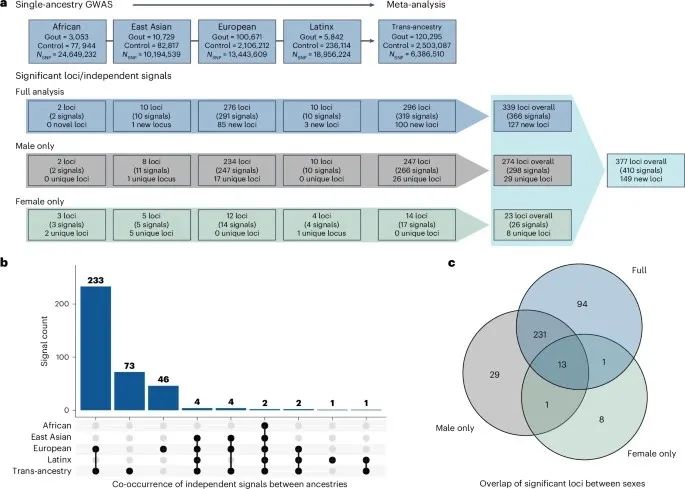
图1:痛风位点分析
此外,研究使用欧洲祖源群体的数据开发了一个痛风多基因风险评分(PRS),显著提高了对痛风风险的预测能力——UK Biobank的参与者根据其PRS值分为10组,痛风患病率从0.0%增加到22.6%。
为了深入了解候选因果基因,本研究选择了608个候选因果SNP。在具有变异功能影响的16个基因中,有7个先前未报道与痛风有关。本研究确定了9个非常强的候选因果变异,这些变异包括ABCG2、CPS1和FGF21的错义变异以及其他6个位点的候选调节变异。
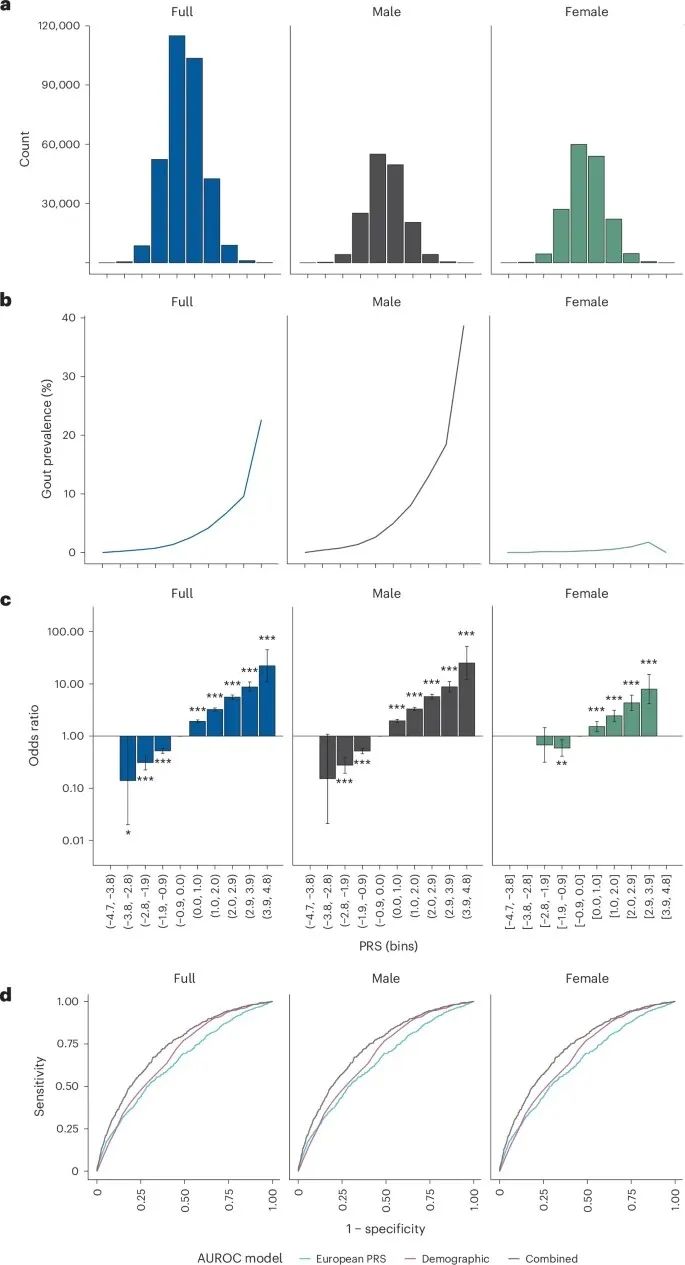
图2:PRS构建
单核苷酸多态性(single nucleotide polymorphism,SNP)主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性。由单个碱基的转换或颠换所引起,也可由碱基的插入或缺失所致。
通俗一点来讲就是,我们人类的基因中大概有99%的基因都是相似的,但是我们每个人所表现出的外貌、体型等都各不相同,这就是因为在DNA片段中有单个核苷酸或单个碱基发生了突变,即存在一些可变的位点,这些可变的位点大概占我们全部遗传信息总量的1%。
2、痛风遗传控制与尿酸的关系
这项研究使用LDSC来研究欧洲痛风GWAS与英国生物银行934个性状之间的遗传相关性,结果显示:痛风与348种表型之间存在遗传相关性,与尿酸的相关性最强,覆盖了27个宽泛表型类别中的25个。
在这些类别之外,与性别相关的两个血液生物标志物(睾酮和性激素结合球蛋白)与痛风显示出负相关,这与最近的一项报告一致,即这些激素的遗传预测水平越高,痛风风险越低。痛风与白细胞、淋巴细胞、中性粒细胞、嗜酸性粒细胞和网织红细胞的血液计数显示出正遗传相关性。在欧洲英国生物银行参与者中,尿酸水平的遗传相关性类似,包括性别特定的分析,348个与痛风相关的表型中有300个也与尿酸显著相关,并且方向一致。
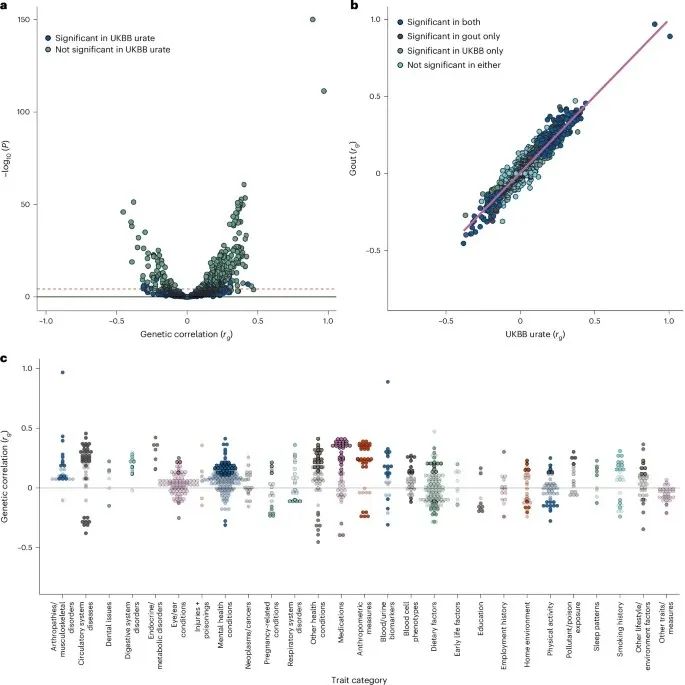
图3:遗传相关性的火山图
在GWAS分析中我们可以通过协变量来校正群体分层等因素,但无法完全消除混杂因素的影响,这时我们可以通过LDSC分析来探究混杂因素的占比,从而检验分析结果的可靠性。
LDSC(LD score regression)是一种常用的遗传相关性分析方法,用于估计复杂性疾病和复杂性特征的遗传贡献。基于遗传连锁不平衡(LD)的概念,通过估计每个SNP的LD Score来推断其与复杂性状之间的关联强度。其本质就是一个线性回归,将GWAS分析结果作为LDSC的分析数据。
3、候选基因、组织和通路
采用协变量调整后的LDSC来分析祖先群体数据中组织和细胞类型的富集情况。结果发现,肾脏和肝脏组织在两人群中均显著富集,且在欧洲人群中发现了与肠道相关的细胞类型富集。
这项研究发现了与痛风相关的组织特异性eQTL基因,如在睾丸中与尿酸稳态相关的HNF4G和PRPS2基因。此外,血液DNA甲基化分析显示痛风位点可能控制着DNA甲基化状态,涉及27个转录因子的结合位点。最终,研究确定了104条生物学通路的富集,包括尿酸代谢和染色质修饰,其中20个组蛋白基因在多个通路中起关键作用。
eQTL共定位分析
GWAS分析找到显著信号位点后,需要解释显著信号位点是如何影响表型,常见的一个解释方法就是共定位分析。
当检测到GWAS信号和eQTL共定位时,我们会认为GWAS信号上的位点可能通过改变基因表达的生物学过程从而影响表型。
4、痛风相关的炎症机制
基于5,358个独特基因的数据,通过GWAS定位、cis-eQTL和trans-eQTL基因分析,优先考虑了108个可能涉及痛风炎症反应的基因。
排序最高的基因包括与脂质代谢有关的FADS1、DGAT2和FADS2,以及与炎症反应相关的IRF5、IL1R1、TRAF4、IL6R和MAST3。为了鉴定候选IPL,这项研究鉴定了76个与痛风GWAS信号共定位的lncRNA物种的61个顺式eQTL,占所有共定位的顺式eQTL基因的18%。
然后,本研究根据全血中具有eQTL且优先级评分高于4,确定了5个候选IPL位点。2个IPL在全血中具有eQTL(DGAT2, RP11-535A19.2和BAG4, RP11-350N15.5),对DGAT2位点的检测表明,该区域受DNA甲基化和RP11-535A19.2表达介导的免疫细胞特异性调控。
此外,通过Genome for Repositioning pipeline系统性地识别了所有GWAS位点中的药物靶点,显著类别包括肿瘤、血液生物化学和代谢紊乱,主要由CASP9、CCND1、CHEK2、DRD5、IGF1R、INSR、PPARG和DGAT2驱动。
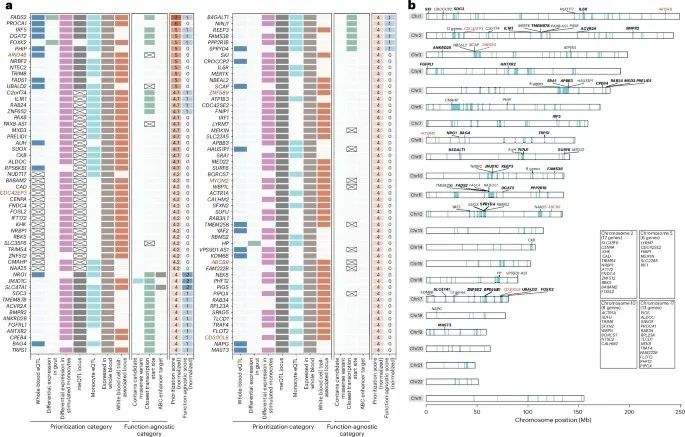
图4:在痛风炎症中发挥作用的优先基因
二、小结
这项研究通过精准的全基因组关联分析(GWAS),在260万人中识别出377个与痛风相关的基因位点,其中包括410个遗传独立的信号,其中有149个位点是之前未被报道过的。研究团队不仅在欧洲人群中发现了这些位点,还在非洲、东亚和拉丁美洲人群中进行了验证。这种跨种族的全面分析策略,不仅增强了研究结果的普适性和可靠性,还为痛风的遗传学研究提供了宝贵的资源。
此外,研究团队运用前沿技术,如基因表达量性状位点(eQTL)分析和DNA甲基化QTL(meQTL)分析,成功地将基础研究与临床应用紧密联系起来,为痛风的治疗提供了新的策略,显示了跨学科研究方法在提高研究创新性和临床相关性方面的巨大潜力。
参考文献:
[1] Major TJ, Takei R, Matsuo H, et al. A genome-wide association analysis reveals new pathogenic pathways in gout. Nat Genet. Published online October 15, 2024. doi:10.1038/s41588-024-01921-5
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言











#痛风#的内因是基因层面的问题,包括基因表达,甚至基因突变的异常。但是表象是#海鲜##啤酒#等#高嘌呤饮食#是诱因,二者相辅相成。人体#尿酸#形成是自我保护作用,但是生成--排出不平衡就会导致尿酸的沉积,带来痛风,以及#高尿酸血症#,#尿酸性肾病#等一系列问题。 #中医#上一般采用#补气#(#补气化浊#)#祛湿#(#芳香化湿##清热利湿#)等方法结合,实际上,通过这些药物调节与尿酸代谢相关的基因表达,以及肾小管对尿酸的排泄作用。中药的作用相对微弱,但是作用是多方面的,西药,如#秋水仙碱#,#非布司他#等,更具有针对性,但是作用是单一的。
3
#全基因组关联研究# #痛风#
5