
Nat Commun发表迄今最大的亚洲多祖先SV图谱,揭示亚洲人群的基因组变异模式
昨天 测序中国 测序中国 发表于上海

新加坡团队利用亚洲血统新加坡人 WGS 数据开展大规模 SV 研究,分析不同祖先群体 SV 图谱、特性、影响等,创建亚洲最大 SV 数据库,填补亚洲人群基因组变异理解空白,有重要价值。
近年来,结构变异(SV)已成为重要变异来源之一。随着WGS可用性和SV调用算法的发展,研究人员越来越多地利用短读长WGS数据来表征人类SV图谱。然而,目前使用WGS的大规模SV研究主要集中在欧洲人群,对其他祖先群体尤其是亚洲人的SV多样性知之甚少。新加坡是由印度裔、华裔和马来裔个体组成的多种族国家,可作为东亚、东南亚和南亚人群的一个缩影,适用于分析亚洲人群SV图谱和基因组变异。
基于此,新加坡科技研究局联合多单位研究人员报告了首个大规模亚洲人群多祖先SV研究,利用来自8392名亚洲血统新加坡人(SG10K_Health)的WGS数据和专门的SV-calling工具鉴定并表征了人群中的SV,并将这些SV与调控和生物学效应相关联,填补了解读亚洲人群基因组变异图谱的关键空白,对理解亚洲人群的基因组变异模式具有重要意义。该项工作以“A Catalogue of Structural Variation acrossAncestrally Diverse Asian Genomes”为题发表在Nature Communications上。

1 新加坡三个主要祖先群体的SV图谱
研究团队分析了来自SG10K_Health研究9770个样本的Illumina短读长WGS数据,包含华裔(58%)、印度裔(24%)和马来裔(18%)参与者。经质控筛选后,得到8392个样本组成的SG10K-SV-r1.4数据集。为了提高SV分析的准确性并减少技术误差,研究人员将其分为三个不同的数据集,即(1)包含5487名个体的发现队列(平均测序深度:15×,文库构建方法:PCR +),(2)包含1523名个体的15x验证队列(平均测序深度:15×,文库构建方法:PCR - ),(3)包含1922名个体的30x验证队列(平均测序深度:30×,文库构建方法:PCR +),重点关注发现队列以识别SV,另两个验证队列用于确保结果可重复性。总体而言,该研究代表了迄今为止最大规模的亚洲SV研究之一(图1a),涵盖的亚洲血统个体数量是先前研究的4倍,且包含此前未研究的东南亚血统群体。
对于SG10K - SV - r1.4发现数据集,研究团队聚焦于三种最常见的SV类型:缺失、插入和重复(图1b)。由于使用单一分析工具难以准确识别SV,研究人员对多个SV检测工具进行基准测试(包括Manta、Delly和Smoove),以确定生成SG10K - SV图谱的工具。结果显示,Delly的精确率优于Manta,但Manta的总体F1得分高于其他工具单独或组合使用(图1c - e),研究还估计了在不同测序深度下SV检测流程可能遗漏的SV比例,发现Manta在检测含串联重复序列区域(如微卫星和小卫星)的重复方面存在局限性,研究使用SurVIndel2对Manta进行了补充。
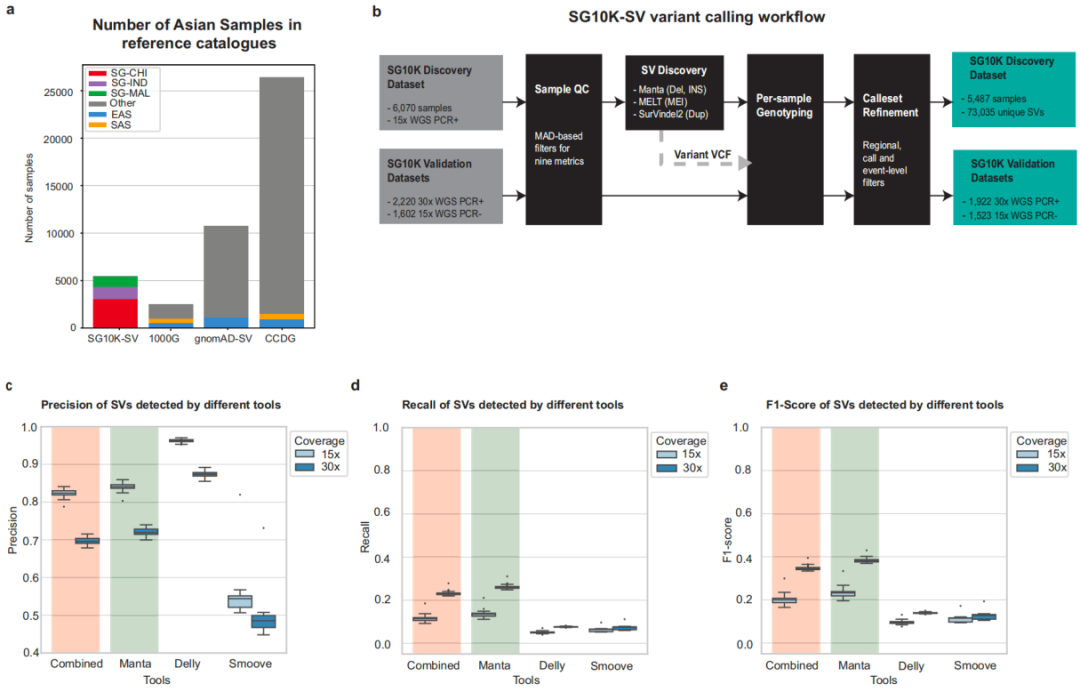
图1. SG10K-SV-r1.4 SV图谱
类似地,移动元件插入(MEIs)呈现出MELT算法具有卓越检测能力的特性。综合使用这三种工具,研究识别了73,035个SV,包括29,011个插入(包括MEI)、11,560个缺失和32,464个重复。与现有的gnomAD-SV和1000基因组计划3期SV(1000G-SV)相比,SG10K-SV-r1.4事件中大约有66.5%和86.7%是新发现的(图2a,b),体现了分析亚洲基因组的潜力。此外,研究团队利用发现数据集中的变异在两个验证数据集中进行了基因分型,以确保在发现数据集中观察到的结果可重复,为后续遗传学研究提供了可靠的数据基础。
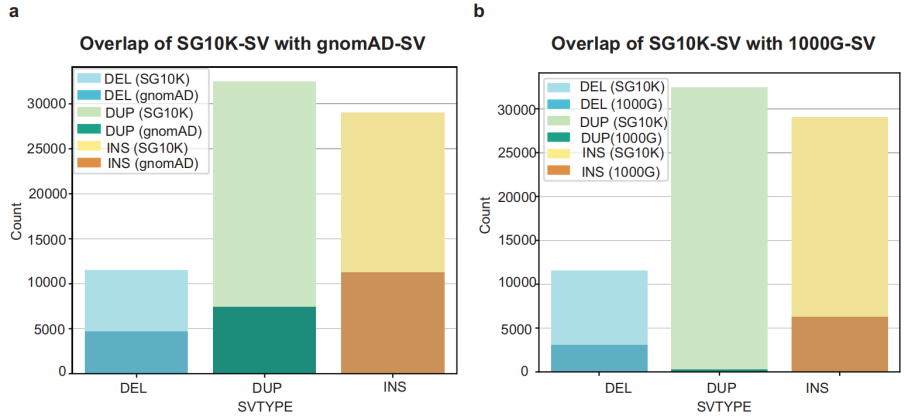
图2. SG10K-SV-r1.4新发现的变异数量
2 SG10K_Health SV特性分析
分析显示,SG10K_Health个体平均含有1439个插入、1584个缺失和1103个重复,在三个祖先群体保持一致(图 3c)。与gnomAD-SV相比,SG10K_Health中检测到的插入和缺失数量较少,这可能是由于gnomAD-SV的测序深度更高;在30×验证数据集中检测到的插入和缺失数量与gnomAD-SV相当。虽然发现队列测序深度较低,但重复数量与gnomAD-SV相当,这可能归因于SurVIndel2重复检测流程的改进。与之前的研究类似,大多数SV的删除、插入和重复是罕见事件,具有低等位基因频率(AF≤1%)(图 3d)。同时,发现队列中也确定了465个等位基因频率大于0.95的SV,表明参考基因组携带次要等位基因。
大多数检测到的SV都较小,但确定了一些长度大于10kb的缺失和重复,在300bp、2kb和6kb处有大量SV(图 3c),其中300bp和6kb的插入分别对应于人类基因组中最丰富的两类转座元件Alu和LINE1。2kb的SV代表复合SVA转座子。SV通常聚集在特定基因组区域,即热点区域。为了识别SV热点区域,研究人员使用hotspotter确定了251个包含高于预期SV密度的区域,其中36%位于染色体末端或着丝粒附近,排除这些区域后,与gnomAD-SV相比,SG10K-SV有88个独特的热点区域。
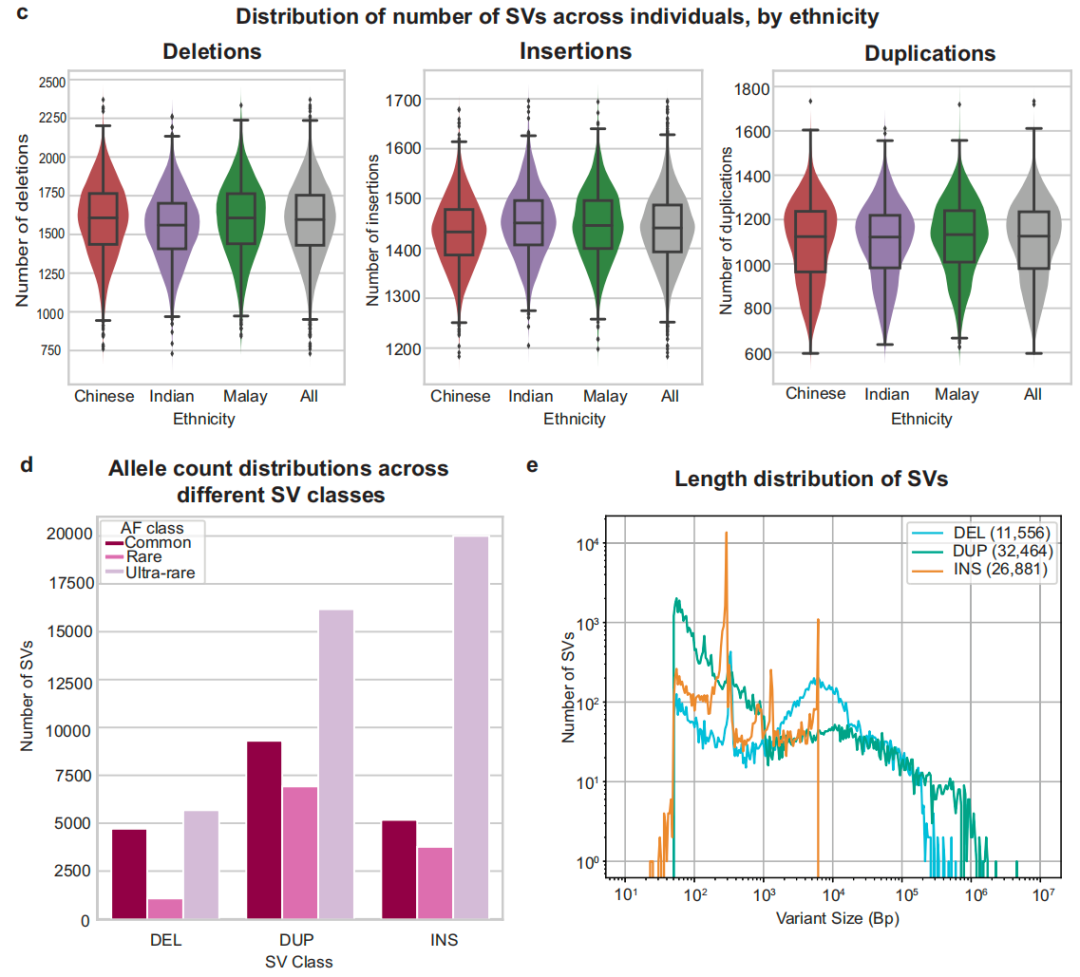
图3. SG10K-SV-r1.4结构变体图谱属性
3 SV对调控元件和基因体的影响
为了评估SG10K-SV对不同功能基因组区域的影响,研究团队将SV与由ENCODE计划和表观基因组学路线图项目确定的基因调控元件进行了重叠分析。结果显示,常见缺失在假定增强子、绝缘子处显著减少,符合负选择模型,罕见及极罕见缺失未表现出类似的信号减少。常见重复在远端和近端增强子处显著减少(图4a),且在非启动子H3K4me3区域富集,推测可能与映射伪影有关。
随后,研究人员对基因体的组成部分(如非翻译区、编码区、外显子、内含子等)进行分析,发现三类SV在基因体各部分普遍减少(图4b)。删除基因区域的SV可致功能丧失,基因重复可能导致基因拷贝数增加,从而增加基因剂量。研究人员通过SVTK43评估SG10K SV对蛋白质编码区域影响,鉴定出2153个SV对蛋白质编码完整性有直接预测影响(图4c),包括导致基因功能丧失及拷贝数增加的情况,这些模式与gnomAD-SV中的发现一致。此外,研究还评估了SV对主要临床可操作基因的潜在影响,在11个临床可操作的ACMG基因中发现了14个影响编码序列完整性的SV(图4d)。
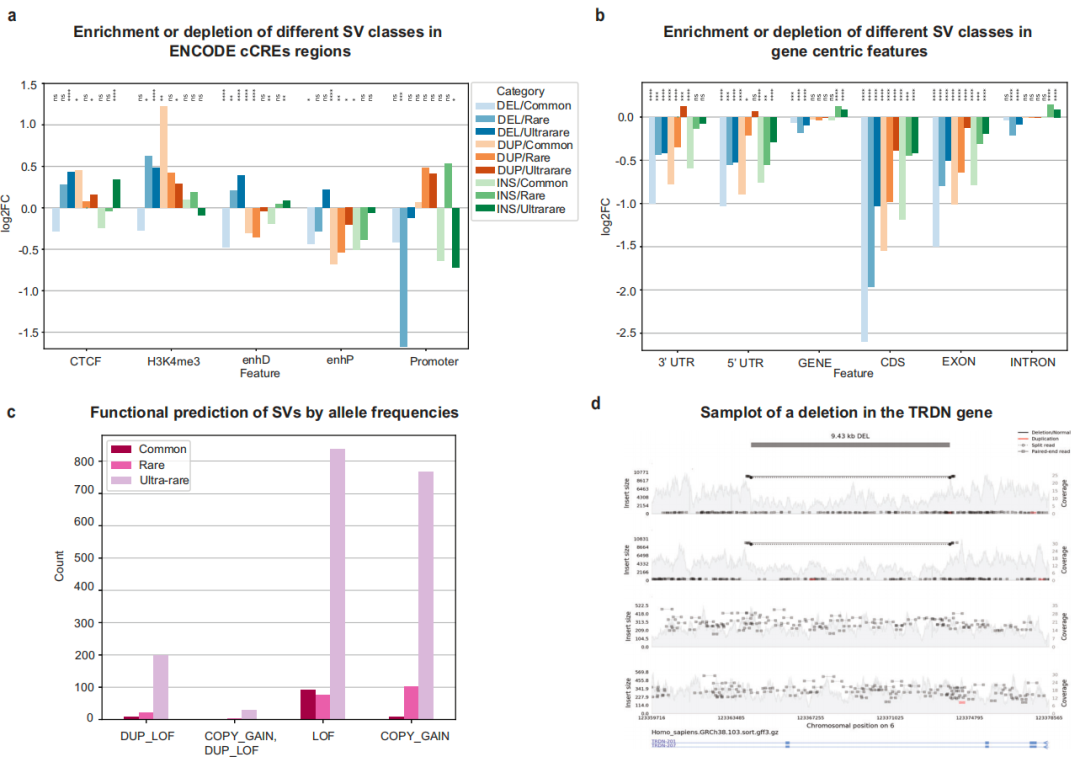
图4. SG10K-SV对功能的影响
4 国际队列间的SV模式
SG10K-SV图谱鉴定的73,035个SV中,分别有66.5%和86.7%的SV未在gnomAD-SV或1000G-SV图谱中报道过。SG10K-SV中有47,770个SV与这两项研究(1000G-SV和gnomAD-SV)无重叠。此外,在SG10K-SV中,对每个族群应用检出率临界值≥50%的标准,鉴定出了42,239个SV,即亚洲特异性新SV。聚焦SG10K-SV中与至少一项研究有重叠的25265个SV,通过固定指数(Fst)分析鉴定出另外10,902个亚洲特异性SV。
5 亚洲多祖先群体间的SV模式
随后,研究团队探究了亚洲三大族裔特有的SV模式,对全套SG10K-SV数据进行主成分分析(PCA),发现了祖先特异性的群体聚类(图 5a)。其中52%的SV仅在一个族裔中出现,13%的SV在两个族裔中共享,其余35%的SV在所有三个族裔群体中均存在(图5b)。通过计算Fst值和置换分析,研究识别了11,715个有祖先特异性频率模式的SV,其中86个影响基因拷贝数或功能丧失,这些影响基因完整性、与特定祖先相关的SV在不同人群水平的等位基因频率范围内均有出现(图5c,d)。特别地,研究人员还发现了一些此前未报道过的SV,包括SMC1B基因的942bp重复、ZNF83基因的84bp删除、CEACAM3基因的209bp插入、TRIM48基因的6.9kb删除等,这些SV在中国、印度、马来等不同族裔中的等位基因频率也各有特点。因此,使用SG10K-SV可检测到亚洲人群中众多特异性SV。
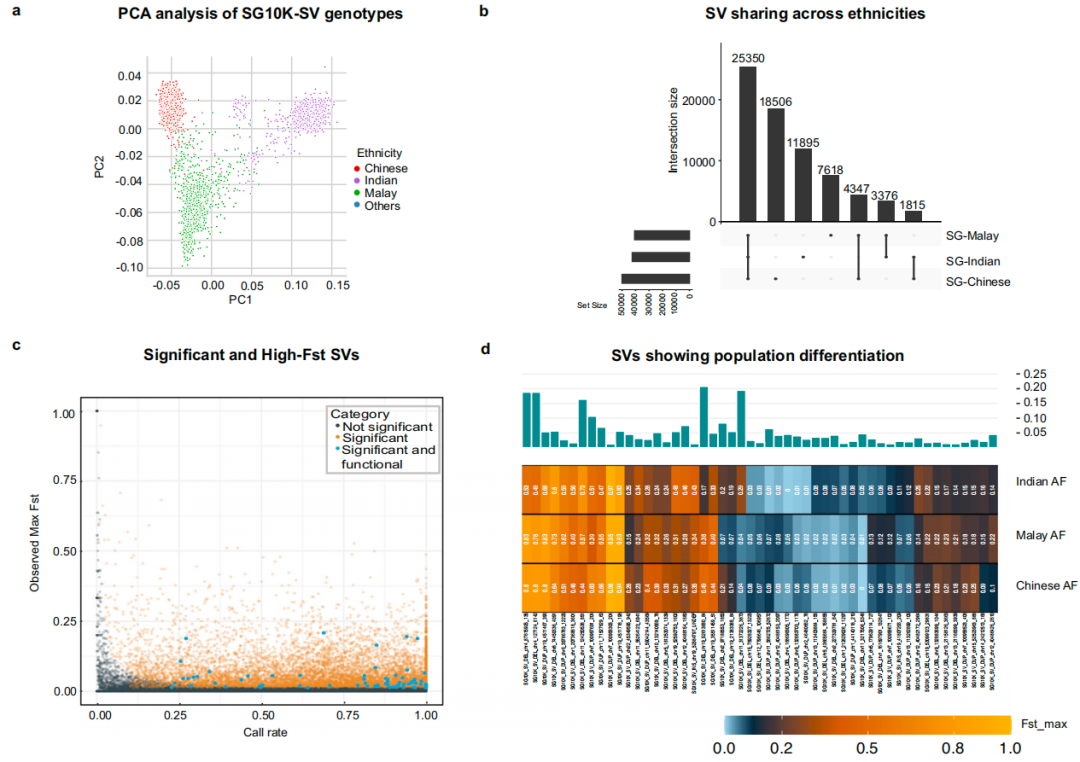
图5. SV的人群特异性
6 SV与GWAS基因位点呈现顺式连锁
SV作为疾病易感性、药物反应及其他表型的潜在遗传驱动因素受到关注。对SG10K-SV和SG10K_Health SNP进行连锁不平衡分析,部分SV与SNP无连锁不平衡,部分有强连锁不平衡,其中一些与GWAS图谱中及针对亚洲队列GWAS的首要SNP有强连锁不平衡,可能是潜在的致病变异。
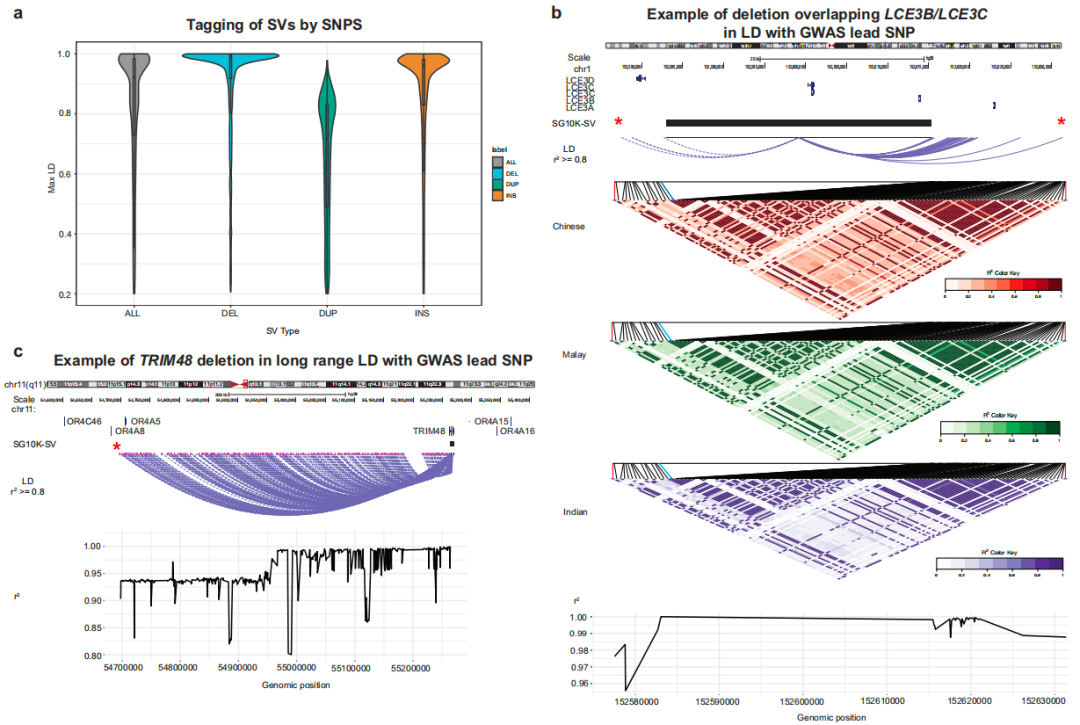
图6. SV和SNP之间的连锁不平衡
综上所述,研究团队借助新加坡多元族裔特点来评估亚洲SV遗传多样性模式,创建了迄今为止最大的亚洲SV数据库,包含73035个SV,其中约65%(47,770个SVs)是新发现的。研究显示,亚洲人群可通过其全球SV模式进行分层,确定了42,239个亚洲人群特有的新型SV,其中52%局限于三个主要祖先群体(印度人、中国人或马来人)之一。此外,研究还发现了影响主要临床可操作位点的SV,并通过鉴定与SNP连锁不平衡的SV,证明了该图谱在亚洲GWAS变异的精细定位和潜在致病变异鉴定中的应用价值。该研究结果填补了对亚洲人群遗传多样性理解的空白,对了解驱动种族群体之间差异的遗传变异及其对不同人群健康疾病状况影响有重要价值。
论文原文:
Tan, J.H.J., Li, Z., Porta, M.G.et al. A Catalogue of Structural Variation across Ancestrally Diverse Asian Genomes. Nat Commun 15, 9507 (2024). https://doi.org/10.1038/s41467-024-53620-8
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言













#基因组# #结构变异#
1