基于中国诊疗现状的《NCCN指南:原发性皮肤淋巴瘤(2024年第1版)》解读
2024-11-01 协和医学杂志 协和医学杂志 发表于上海

本文结合我国原发性皮肤淋巴瘤的诊疗现状与特点,对指南的主要推荐内容进行解读,以帮助读者更好地理解其核心要点。
原发性皮肤淋巴瘤是一类具有特殊临床表现以及分子生物学特征的淋巴瘤,在淋巴瘤分类中作为独立的一组疾病。根据肿瘤细胞来源不同,可分为原发性皮肤T细胞淋巴瘤(CTCL)和原发性皮肤B细胞淋巴瘤(CBCL),前者包括10余种亚型,其中蕈样肉芽肿(MF)、Sézary综合征(SS)及原发性皮肤CD30+T细胞淋巴增殖性疾病较为常见(占比90%)[1],后者主要包括原发性皮肤边缘区淋巴瘤(PCMZL)、原发性皮肤滤泡中心淋巴瘤(PCFCL)和原发性皮肤弥漫性大B细胞淋巴瘤,腿型(leg type,PCDLBCL,LT)3种亚型。
美国国立综合癌症网络(NCCN)是由33家权威癌症中心组成的学术机构,其发布的肿瘤临床实践指南得到全球临床医生的广泛关注。2023年12月,NCCN更新发布了《NCCN指南:原发性皮肤淋巴瘤(2024年第1版)》(下文简称“指南”)[2],该指南基于循证医学证据和最新研究进展提出了针对上述原发性皮肤淋巴瘤主要亚型的诊断、评估、辅助检查、分期及治疗相关建议,为临床医生提供了具有指导意义的规范化诊疗策略。
与2023年版指南比较,此次更新主要集中于诊断评估原则、分期标准、不同分期MF的治疗原则及对放疗的建议等方面。本文结合我国原发性皮肤淋巴瘤的诊疗现状与特点,对指南的主要推荐内容进行解读,以帮助读者更好地理解其核心要点。
1 原发性皮肤T细胞淋巴瘤
1.1 MF/SS
MF是最常见的皮肤T细胞淋巴瘤,大多数患者在早期表现为皮肤红斑、斑块,病情通常进展缓慢,进展期可能出现红皮病、皮肤肿物以及淋巴结、血液或其他脏器受累。SS是一种白血病型皮肤T细胞淋巴瘤,以红皮病和外周血出现异型淋巴细胞(Sézary细胞)为主要特征[3]。
1.1.1 诊断
MF/SS的诊断需根据临床表现、病理检查结果、免疫组化染色和TCR基因重排检测进行综合判断。
1 组织病理检查:指南强调在进行皮肤活检前应至少暂停局部治疗2~3周,以减少治疗对诊断的影响。若病理结果不具有诊断价值或与临床表现不一致,应重复活检。
2 免疫组化染色:常规包括CD2、CD3、CD4、CD5、CD7、CD8、CD20、CD30。对于诊断困难的患者,还可进行CD25、CD56、TIA1、颗粒酶B、TCR-β、TCR-δ、CCR4、CXCL13、ICOS、PD-1检测。MF/SS肿瘤细胞典型的免疫表型为CD2+、CD3+、CD4+、CD5+、CD7-、CD8-,但少数情况下可见CD8+变异型。发生大细胞(超过25%的肿瘤细胞为大细胞)转化时可见CD30+。
3 TCR基因重排检测:检测到TCR基因克隆性重排有助于明确MF/SS诊断,可辅助鉴别MF/SS与炎症性疾病。
4 外周血流式细胞分析:若皮损广泛而病理表现不具有诊断意义,应进行外周血Sézary细胞检测。
1.1.2 辅助检查
查体时应对患者的皮损类型(斑片、斑块、肿瘤或红皮病)和所占的体表面积进行评估。此外,应进行浅表淋巴结和腹部触诊,以评估是否存在淋巴结肿大或肝脾肿大,对于可疑皮肤外受累的部位,应进行手术活检或穿刺活检。如活检结果不具有诊断意义或与临床表现不一致,则应重复进行活检。指南新增了“活检结果与临床表现不一致”这一重复活检指征。
实验室检查应包括血常规、包含乳酸脱氢酶(LDH)在内的生化检查、TCR基因重排检测,T2~T4期或怀疑皮肤外受累的患者还应进行流式细胞分析以检测异常表型的T细胞亚群。
影像学检查推荐胸腹盆增强CT或PET/CT。对于分期≥T2b(表1)、亲毛囊性MF、存在大细胞转化、可触及淋巴结肿大或实验室检查结果异常的患者,应进行PET/CT检查。
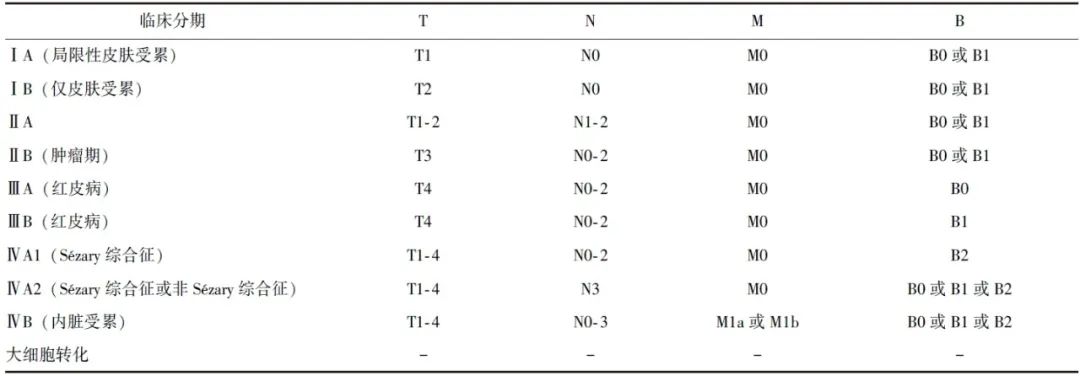
表1 MF/SS的TNMB分期


1.1.3 分期
采用TNMB分期方法对MF/SS进行分期[4],在淋巴结、血液及脏器受累情况的评估方面,与上一版指南相比,指南根据肿瘤细胞的克隆性与皮肤是否一致以及受累脏器的类型进一步细化了分期规则(表1)。
对于皮肤(T)分期,指南强调,如在初始评估时分期为T4,在治疗期间皮损面积缩小并不改变分期。在评估血液受累情况时,淋巴细胞减少症(淋巴细胞数目<1000/μL)患者应通过异常淋巴细胞数目和异常淋巴细胞百分比进行评估,以避免低估异常淋巴细胞负荷。
MF/SS的临床分期(表2)[4]由TNMB分期决定且与治疗方案的选择密切相关。大细胞转化是一种组织学特征,在各分期均可能出现,由于发生大细胞转化的患者通常需要更为积极的治疗,因此指南在临床分期系统中将大细胞转化作为独立分类,并单独给出治疗建议。
表2 MF/SS临床分期系统
1.1.4 治疗
1.1.4.1 治疗原则
MF/SS的治疗以缓解症状、控制病情、延缓进展为目标。目前大多数治疗方法在停止治疗后不能获得持续缓解,除异基因造血干细胞移植外,其他疗法均无法治愈,治疗方案详见图1。
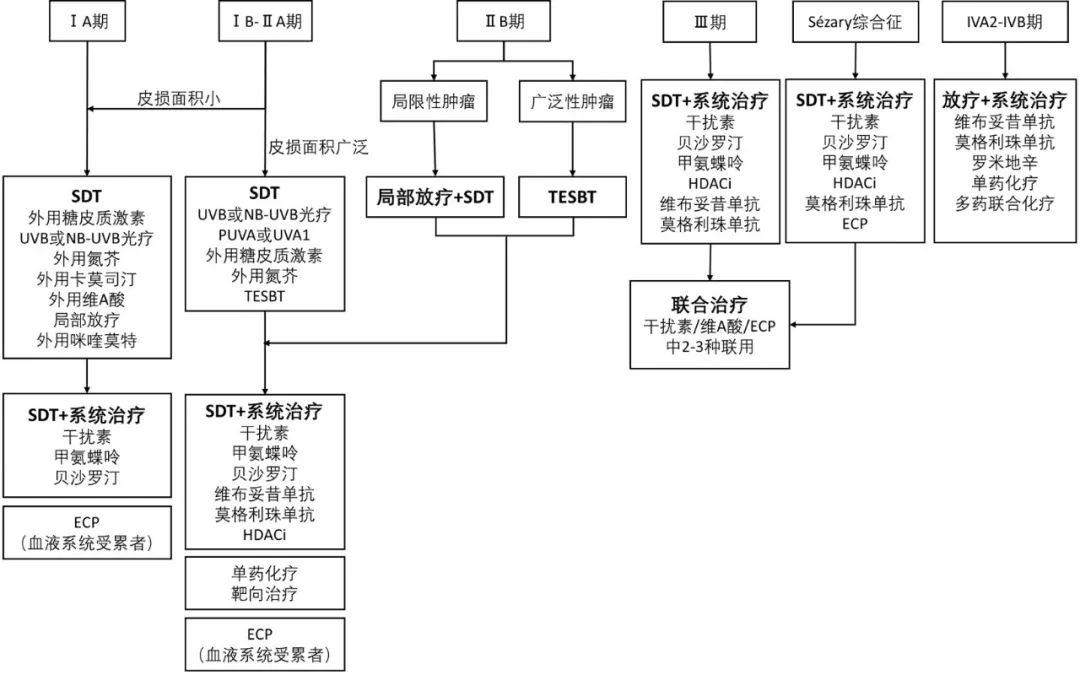
图1 MF/SS治疗方案
SDT( skin-directed therapy):皮肤定向治疗;UVB(ultraviolet B):中波紫外线;NB-UVB(narrow bound ultraviolet B):窄谱中波紫外线;ECP(extracorporeal photopheresis):体外光化学疗法;PUVA(psoralen plus ultraviolet A):补骨脂素长波紫外线;UVA1(ultraviolet A1):长波紫外线;TSEBT(total skin electron beam therapy):全身皮肤电子束疗法;HDACi(histone deacetylase inhibitors):组蛋白去乙酰化酶抑制剂
在选择治疗方案时,应根据患者的症状、分期、耐受性及治疗目标制订个体化方案。分期Ⅳ期、年龄>60岁、存在大细胞转化以及LDH升高是MF/SS预后不良的独立危险因素,应给予更为积极的治疗[5]。初始治疗建议首选皮肤定向治疗(SDT)和耐受性良好、无累积毒性、免疫抑制程度较轻和/或疗效较好的系统治疗,需化疗的患者应首选单药化疗。系统治疗可联合SDT以提高疗效。
1.1.4.2 不同分期的治疗方案
1 ⅠA期
指南建议ⅠA期患者首选SDT,可单独使用或多种SDT联合应用。推荐的治疗方案包括:
1 外用糖皮质激素
在外用糖皮质激素时,应关注皮损部位和使用时间,薄嫩皮肤长期使用强效糖皮质激素可能导致全身吸收和皮肤萎缩等不良反应。口周、眶周等部位的皮损,可使用钙调磷酸酶抑制剂(如吡美莫司)代替。
2 全身紫外线B(UVB)或窄谱紫外线B
(NB-UVB)光疗
根据美国皮肤淋巴瘤协会的建议,UVB起始治疗频率为每周3次,皮损完全消退后应维持当前治疗频率及剂量治疗1~3个月,此后可缓慢降低治疗频率直至停止治疗,建议该阶段应至少持续3个月[6]。
3 外用氮芥
氮芥凝胶已被美国食品药品监督管理局(FDA)批准用于早期MF的局部治疗,临床研究显示,氮芥凝胶的治疗有效率为59%[7]。指南对氮芥的使用方法进行了更新,由于氮芥可能引起皮肤刺激症状,初始使用频率应低于每日1次,可联合外用糖皮质激素以减轻刺激症状。氮芥可单独使用或与其他SDT联合使用,如与光疗联合治疗,应在照光后使用氮芥。
4 外用卡莫司汀
卡莫司汀可用于斑片或斑块期MF的治疗,一项临床研究显示,卡莫司汀治疗T1期MF的有效率为92%[8]。卡莫司汀对局部皮肤的副作用较小,但可能出现全身吸收,导致骨髓抑制的风险增加。
5 外用维A酸类药物
指南推荐的外用维A酸类药物包括贝沙罗汀和他扎罗汀。贝沙罗汀凝胶是唯一被FDA批准用于MF/SS的外用维A酸类药物,治疗早期MF的总体有效率为63%[9]。维A酸类药物可能引起红斑、脱屑等皮肤刺激症状,应尽量避免用于间擦部位及面部。
6 局部放疗
常用剂量为8~12 Gy,局部低剂量放疗是缓解病情的手段之一,常与其他治疗联合使用。
7 外用咪喹莫特
咪喹莫特推荐小范围用于较顽固的皮损。
对于SDT治疗效果不佳的患者,可使用系统治疗联合SDT,该阶段患者的系统治疗以免疫调节类药物为主,指南推荐的治疗方案包括:
1 干扰素(IFN)
IFN包括IFN-α、IFN-β、IFN-γ,在MF的治疗中以IFN-α最为常用,皮下注射起始剂量通常为300万单位/次,每周3次,根据治疗反应可逐渐增加或减少剂量[10]。当IFN-α不可用时,指南建议选择IFN-γ-1b。
2 甲氨蝶呤(MTX)
低剂量MTX是MF系统治疗中最常用的单药化疗药物,低剂量MTX副作用相对较小,可作为初始系统治疗选择,常用剂量为5~25 mg/次,每周1次[10]。
3 贝沙罗汀
贝沙罗汀属于维A酸类药物,在一项针对难治性早期CTCL患者的临床试验中,口服贝沙罗汀每日300 mg/m2 的整体缓解率为54%[11]。目前贝沙罗汀在国内尚未上市,可使用阿维A或异维A酸替代。
4 体外光化学疗法(ECP)
ECP可用于在早期出现血液受累(B1期)的患者。
2 ⅠB~ⅡA期
ⅠB~ⅡA期患者的治疗以SDT为主。对于皮损面积较小或以斑片为主的患者,治疗选择与ⅠA期类似。对于应答不充分(未达到部分缓解或全部缓解)的患者,上一版指南建议按照皮肤肿瘤负荷较重的情况进行治疗,新版指南新增了再次尝试初始治疗方案的建议。
如皮损面积广泛或以斑块为主,指南建议首选的治疗方案包括:
|
1 |
全身UVB或NB-UVB光疗; |
|
2 |
补骨脂素长波紫外线(PUVA)或长波紫外线(UVA1)光疗; |
|
3 |
外用糖皮质激素; |
|
4 |
外用氮芥; |
|
5 |
全身皮肤电子束疗法(TSEBT)。 |
在进行光疗时,应注意紫外线的累及剂量与部分皮肤肿瘤的发病风险呈正相关,PUVA的风险相对更高,在治疗前应评估患者的风险与获益。
对于皮肤肿瘤负荷较重、有血液系统受累或对SDT治疗反应不佳的患者,可联合使用系统治疗。
首选的治疗方案除ⅠA期推荐的系统治疗方案外,还包括:
1 维布妥昔单抗(BV)
BV是一种以CD30为靶点的抗体偶联药物,一项Ⅱ期临床研究显示,28例接受BV治疗的MF患者中,总体有效率为54%[12]。BV已被批准用于接受过系统治疗的CD30+MF成年患者。
2 莫格利珠单抗(mogamulizumab)
莫格利珠单抗以CCR4为治疗靶点,已被批准用于接受过至少1次系统治疗的复发或难治性MF/SS成年患者。一项Ⅲ期临床试验结果显示,接受莫格利珠单抗治疗的MF/SS患者中位无进展生存期为7.7个月[13]。
3 组蛋白去乙酰化酶抑制剂(HDACi)
指南推荐的HDACi包括罗米地辛和伏立诺他,但均未在国内上市,目前我国自主研发的西达本胺可供临床使用,已被批准用于接受过至少1次全身化疗的复发或难治性外周T细胞淋巴瘤患者。
4 化疗
对上述系统治疗反应不佳的患者,可考虑单药化疗,常用方案包括吉西他滨、多柔比星脂质体和普拉曲沙。
5 其他靶向治疗药物
指南推荐的其他靶向药物包括阿仑单抗和帕博利珠单抗。阿仑单抗是一种针对CD52的人源化IgG1抗体,一项针对Ⅲ~Ⅳ期MF/SS患者的临床研究显示,总体有效率为55%[14],阿仑单抗目前尚未在国内上市。帕博利珠单抗是一种抗PD-1免疫检查点抑制剂,24例接受帕博利珠单抗治疗的ⅡB~ⅣB期MF/SS患者中,总体有效率为38%[15]。帕博利珠单抗已在国内上市,但尚未获批用于MF/SS的治疗。
3 ⅡB期
1 局限性肿瘤
该阶段的患者均推荐局部放疗联合SDT,可根据病情选择是否同时进行系统治疗。指南强调了SDT的治疗地位,无论是否进行系统治疗,均推荐使用SDT。系统治疗的选择与ⅠB~ⅡA期类似,但不推荐单药化疗。
2 泛发性肿瘤
对于肿瘤泛发的患者,可进行TSEBT、系统治疗联合SDT或多种系统治疗联合使用。系统治疗方案与ⅠB~ⅡA期类似,但对单药化疗的推荐等级更高。联合使用多种系统治疗时,应首选不良反应较小的治疗方案,指南推荐口服维A酸类药物联合IFN-α;对于有血液系统受累的患者,可同时联合使用ECP。
4 Ⅲ期
Ⅲ期患者临床表现为红皮病,应行系统治疗联合SDT。系统治疗的选择方面,指南推荐首选贝沙罗汀、IFN-α、MTX、罗米地辛、BV、莫格利珠单抗和ECP。此外,还可选择IFN-α、维A酸类药物、ECP中的2~3种联合使用。SDT可选择光疗或TSEBT,由于红皮病患者皮损广泛,光疗和TSEBT引起不良反应的风险增加,在治疗时应注意调整剂量。同时,由于该阶段患者继发感染的风险增加,必要时可联合使用系统性抗生素。
5 SS ⅣA1期或ⅣA2期
SS治疗的基本原则是系统治疗联合SDT。对于Sézary细胞数目<5000/mm3 的患者,首选的治疗方案包括贝沙罗汀、IFN-α、MTX、罗米地辛、伏立诺他、莫格利珠单抗、ECP或联合治疗。对于肿瘤负荷较重(Sézary细胞数目>5000/mm3)的患者,指南推荐首选莫格利珠单抗、罗米地辛或上述联合治疗方案。
6 非SS ⅣA2期和ⅣB期
指南推荐系统治疗联合局部放疗。首选的系统治疗方案包括BV、罗米地辛、吉西他滨、多柔比星、普拉曲沙;此外,可选择莫格利珠单抗或按照外周T细胞淋巴瘤,非特指型进行多药联合化疗。多药联合化疗建议用于复发/难治性或存在皮肤外受累的患者,治疗前建议首先尝试多种单药治疗方案。
7 MF伴大细胞转化
出现大细胞转化的患者通常病情进展迅速,需要更为积极的治疗。指南推荐的首选治疗方案包括TSEBT或系统治疗联合SDT,可选择的系统治疗方案包括BV、罗米地辛、单药化疗(吉西他滨、多柔比星、普拉曲沙)或多药联合化疗。此外,还可尝试帕博利珠单抗治疗。
8 复发难治性MF
对于多种治疗均反应不佳的患者,指南建议患者参与临床试验、评估造血干细胞移植可行性或尝试其他化疗或靶向治疗方案,如阿仑单抗、帕博利珠单抗、苯丁酸氮芥、环磷酰胺、依托泊苷、喷司他丁、硼替佐米、替莫唑胺(中枢神经系统受累患者)以及多药联合化疗。
1.2 原发性皮肤CD30+T细胞淋巴增殖性疾病
原发性皮肤CD30+T细胞淋巴增殖性疾病包括原发性皮肤间变性大细胞淋巴瘤(PC-ALCL)、淋巴瘤样丘疹病(LyP)以及与二者临床和组织病理特征均有重叠的交界性病变。
1.2.1 概述
PC-ALCL临床表现为孤立性结节或肿物,约20%的病例表现为多发皮损,在组织学上以弥漫性CD30+ 大细胞浸润为特征。该病皮肤外受累少见,主要累及区域淋巴结,整体预后良好。LyP临床表现为慢性、复发性、可自发消退的丘疹、结节,皮损通常成群出现。本病组织学表现具有异质性,可见大的间变细胞、霍奇金样细胞和多种炎症细胞混合浸润,根据组织学特点可分为不同亚型。
1.2.2 诊断与辅助检查
诊断需临床与病理相结合并除外其他表达CD30的良恶性疾病,如系统性T细胞淋巴瘤、MF伴大细胞转化、节肢动物叮咬、淋巴瘤样药物反应等。
1 组织病理检查
与上一版指南相比,该版指南强调重复活检和充分取材的重要性。推荐进行环钻活检、切除活检或切取活检,如皮损类型多样,应对所有类型的皮损均进行组织病理检查。
2 免疫组织化学染色
常规包括:CD3、CD4、CD8、CD20、CD30、CD56、ALK。如与其他疾病鉴别困难,可进一步完善CD2、CD5、CD7、CD25、TIA1、颗粒酶B、穿孔素、IRF4/MUM1、EMA、TCR-β、TCR-δ等检测。
3 分子遗传学检测
对于诊断困难的病例, EBER原位杂交、荧光原位杂交检测(ALK和DUSP22基因)以及TCR基因重排检测有助于与其他良恶性疾病相鉴别。
4 系统检查
患者应常规进行血常规、生化检查(包括LDH);对于PC-ALCL患者或临床表现不典型的LyP患者还应进行胸腹盆CT或PET/CT检查。出现淋巴结肿大时应进行淋巴结活检,存在皮肤外受累的患者必要时可进行骨髓活检。
1.2.3 治疗
1 PC-ALCL
如皮损范围局限,指南推荐首选局部放疗或手术切除,皮损复发时可重复上述治疗。一项多中心回顾性研究显示,56例接受放疗(初始治疗或手术后放疗)的PC-ALCL患者中,临床完全缓解率达95%[16]。如皮损分布广泛或多次复发,可选择BV、小剂量MTX(≤50 mg/周)、维A酸、普拉曲沙、IFN等治疗方案。
对于伴有区域淋巴结受累的患者,需要更为积极的治疗。指南推荐方案包括:
1 局部放疗联合BV、MTX、普拉曲沙、CHOP(环磷酰胺+多柔比星+长春新碱+泼尼松)或CHOEP(环磷酰胺+多柔比星+依托泊苷+长春新碱+泼尼松)化疗;
2 BV联合CHP(环磷酰胺+多柔比星+泼尼松)化疗。对于复发性患者,仍可再次使用初始治疗方案;难治性患者可选择参加临床试验或按照MF伴大细胞转化进行治疗。
2 LyP
LyP可自行消退,治疗以控制症状为主要目的,无症状患者可选择观望疗法。如皮损面积较小,推荐的一线治疗方案包括局部外用糖皮质激素和光疗。大多数LyP患者对外用糖皮质激素和光疗治疗反应较好,但复发十分常见。研究显示,48%的患者外用糖皮质激素或光疗后达到完全缓解,中位无进展生存期为11个月[17]。对于皮损泛发的患者,可选择系统治疗,包括口服MTX(10~35 mg/周)、维A酸。复发难治性患者可选择BV治疗,一项Ⅱ期临床研究显示,12例难治性LyP患者接受BV治疗的有效率为100%,58%达到完全缓解[18]。
2 原发性皮肤B细胞淋巴瘤
2.1 常见亚型
1 PCFCL
PCFCL是最常见的CBCL亚型,主要发生于头皮、面部、躯干,表现为孤立的粉色至紫红色斑块、结节、肿物,通常不伴有破溃,病程缓慢,皮肤外播散罕见。肿瘤细胞通常表达CD20、CD79a和BCL-6,部分表达CD10,BCL-2通常为阴性表达。
2 PCMZL
PCMZL在2022版的国际共识分类(ICC)中被称为原发性皮肤边缘区淋巴增殖性疾病,是第二常见的皮肤B细胞淋巴瘤,皮损通常位于头部、上肢和躯干,表现为单发或多发的红色至紫红色丘疹、结节、斑块或肿物。整体病程缓慢,5年生存率为99%,但约50%的患者复发[2]。肿瘤细胞表达BCL-2、CD20、CD79a,CD10和BCL-6通常为阴性表达。
3 PCDLBCL,LT
该亚型在CBCL中最为少见,典型临床表现为腿部红色至蓝色的斑块或肿物,可形成溃疡,病程侵袭,通常预后不良。该病通常为活化B细胞(ABC)亚型,肿瘤细胞表达BCL-2、CD20、CD79a、IRF-MUM1、FOXP1和MYC,CD10通常为阴性表达。原位荧光杂交可见MYC、BCL6和IGH基因异位。
2.2 诊断与辅助检查
1 组织病理检查
推荐进行环钻活检、切除活检或切取活检,若病理表现不具有诊断意义和/或与临床表现不一致,应重复进行活检。
2 免疫组化检查
常规包括CD3、CD10、CD20、BCL2、BCL6和IRF4/MUM1;如诊断困难,必要时可进行Ki-67、CD5、CD21、CD23、CD43、cyclin D1, κ/λ检测。
3 分子遗传学检查
对于鉴别诊断困难的患者,可进一步完善分子遗传学检查,包括:
①EBER原位杂交;
②荧光原位杂交检测t(14;18)染色体异位,有助于与系统性滤泡性淋巴瘤相鉴别;
③IGH基因重排检测以确定B细胞的克隆性。
4 系统检查
指南建议常规进行血常规、生化(包括LDH)、感染4项、胸腹盆CT或PET/CT检查;如血常规提示淋巴细胞增多,可进行外周血流式细胞分析。PCMZL患者应完善血清蛋白电泳或免疫球蛋白定量。出现不明原因血细胞减少或疑诊其他亚型淋巴瘤的患者可进行骨髓活检。
2.3 分期
皮肤B细胞淋巴瘤的分期采用非MF/SS皮肤淋巴瘤的TNM分期方法[4],具体分期标准见表3。
表3 非MF/SS皮肤淋巴瘤的TNM分期

2.4 治疗
1 PCMZL和PCFCL
对于局限性病灶(T1~T2期),指南推荐首选局部放疗、手术切除或二者联合。一项纳入34例接受放疗的CBCL患者的临床研究显示,PCMZL和PCFCL患者的5年无复发生存率为62%~73%[19]。在放疗剂量的选择方面,指南新增推荐低剂量放疗方案,研究显示,4Gy的放疗剂量对于初治及复发性患者均有效[20]。其他可选择的方案包括皮损内注射糖皮质激素或SDT,外用糖皮质激素、咪喹莫特、氮芥及贝沙罗汀。如病情复发,可选择上述治疗建议中的其他方案。
如皮损广泛(T3期),初始治疗建议包括SDT、局部放疗、皮损内注射糖皮质激素和利妥昔单抗,难治性疾病可尝试其他化学免疫联合疗法。
如出现皮肤外受累,指南建议PCFCL患者按照滤泡性淋巴瘤管理,PCMZL患者按照结内边缘区淋巴瘤管理。
2 PCDLBCL, LT
PCDLBCL, LT患者的预后较差,通常需按照系统性弥漫性大B细胞淋巴瘤进行系统化疗。常用的治疗方案是含蒽环类药物的多药化疗联合利妥昔单抗。一项纳入115例患者的临床研究显示,多药化疗联合利妥昔单抗治疗PCDLBCL, LT的3年生存率为80%,5年生存率为74%[21]。
3 小结
原发性皮肤淋巴瘤是一组异质性疾病,部分亚型的诊断与治疗存在挑战。2023年国内先后发表了《中国蕈样肉芽肿诊疗及管理专家指南》和《蕈样肉芽肿治疗中国专家共识(2023)》,推动了我国MF诊疗与管理的规范化,但作为一组分类复杂的罕见疾病,不同医生对原发性皮肤淋巴瘤的认知仍存在较大差别。
该版指南在诊断方面强调多次活检的重要性,并对分期作出了进一步细化。治疗方面,整体遵循2023版指南的治疗原则,以MF/SS治疗为例,建议根据分期制订治疗方案并首选耐受性良好、副作用较小的药物,更新了低剂量放疗方案,在控制症状的同时减少治疗相关不良反应;同时指南强调SDT的重要性,在各个分期均推荐应用。本指南对该病的诊断、评估和治疗提供了更为精准的建议,有助于指导临床进行规范化治疗和患者管理。
参考文献
[1]Willemze R, Cerroni L, Kempf W, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas[J]. Blood, 2019, 133(16): 1703-1714.
[2]National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines for Primary Cutaneous Lymphomas(Version 1.2024)[DB/OL]. [2024-08-01]. http://www.nccn.org.
[3]Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: lymphoid neoplasms[J]. Leukemia, 2022, 36(7): 1720-1748.
[4]Olsen E A, Whittaker S, Willemze R, et al. Primary cutaneous lymphoma: recommendations for clinical trial design and staging update from the ISCL, USCLC, and EORTC[J]. Blood, 2022, 140(5): 419-437.
[5]Scarisbrick J J, Prince H M, Vermeer M H, et al. Cutaneous lymphoma international consortium study of outcome in advanced stages of mycosis fungoides and Sézary syndrome: effect of specific prognostic markers on survival and development of a prognostic model[J]. J Clin Oncol, 2015, 33(32): 3766-3773.
[6]Olsen E A, Hodak E, Anderson T, et al. Guidelines for phototherapy of mycosis fungoides and Sézary syndrome: a consensus statement of the United States Cutaneous Lymphoma Consortium[J]. J Am Acad Dermatol, 2016, 74(1): 27-58.
[7]Lessin S R, Duvic M, Guitart J, et al. Topical chemother-apy in cutaneous T-cell lymphoma: positive results of a randomized, controlled, multicenter trial testing the efficacy and safety of a novel mechlorethamine, 0.02%, gel in mycosis fungoides[J]. JAMA Dermatol, 2013, 149(1): 25-32.
[8] Zackheim H S. Topical carmustine (BCNU) for patch/plaque mycosis fungoides[J]. Semin Dermatol, 1994, 13(3): 202-206.
[9]Breneman D, Duvic M, Kuzel T, et al. Phase 1 and 2 trial of bexarotene gel for skin-directed treatment of patients with cutaneous T-cell lymphoma[J]. Arch Dermatol, 2002, 138(3): 325-332.
[10]Trautinger F, Eder J, Assaf C, et al. European organisation for research and treatment of cancer consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome-update 2017[J]. Eur J Cancer, 2017, 77: 57-74.
[11]Duvic M, Martin A G, Kim Y, et al. Phase 2 and 3 clinical trial of oral bexarotene (Targretin capsules) for the treatment of refractory or persistent early-stage cutaneous T-cell lymphoma[J]. Arch Dermatol, 2001, 137(5): 581-593.
[12]Duvic M, Tetzlaff M T, Gangar P, et al. Results of a phase Ⅱ trial of brentuximab vedotin for CD30+ cutaneous T-cell lymphoma and lymphomatoid papulosis[J]. J Clin Oncol, 2015, 33(32): 3759-3765.
[13]Kim Y H, Bagot M, Pinter-Brown L, et al. Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): an international, open-label, randomised, controlled phase 3 trial[J]. Lancet Oncol, 2018, 19(9): 1192-1204.
[14]Lundin J, Hagberg H, Repp R, et al. Phase 2 study of alemtuzumab (anti-CD52 monoclonal antibody) in patients with advanced mycosis fungoides/Sezary syndrome[J]. Blood, 2003, 101(11): 4267-4272.
[15]Khodadoust M S, Rook A H, Porcu P, et al. Pembrolizu-mab in relapsed and refractory mycosis fungoides and Sézary syndrome: a multicenter phase Ⅱ study[J]. J Clin Oncol, 2020, 38(1): 20-28.
[16]Million L, Yi E J, Wu F, et al. Radiation therapy for primary cutaneous anaplastic large cell lymphoma: an international lymphoma radiation oncology group multi-institutional experience[J]. Int J Radiat Oncol Biol Phys, 2016, 95(5): 1454-1459.
[17]Fernández-de-Misa R, Hernández-Machín B, Servitje O, et al. First-line treatment in lymphomatoid papulosis: a retrospective multicentre study[J]. Clin Exp Dermatol, 2018, 43(2): 137-143.
[18]Lewis D J, Talpur R, Huen A O, et al. Brentuximab vedotin for patients with refractory lymphomatoid papulosis: an analysis of phase 2 results[J]. JAMA Dermatol, 2017, 153(12): 1302-1306.
[19]Smith B D, Glusac E J, McNiff J M, et al. Primary cutaneous B-cell lymphoma treated with radiotherapy: a comparison of the European Organization for Research and Treatment of Cancer and the WHO classification systems[J]. J Clin Oncol, 2004, 22(4): 634-639.
[20]Neelis K J, Schimmel E C, Vermeer M H, et al. Low-dose palliative radiotherapy for cutaneous B- and T-cell lymphomas[J]. Int J Radiat Oncol Biol Phys, 2009, 74(1): 154-158.
[21]Grange F, Joly P, Barbe C, et al. Improvement of survival in patients with primary cutaneous diffuse large B-cell lymphoma, leg type, in France[J]. JAMA Dermatol, 2014, 150(5): 535-541.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言













#解读# #原发性皮肤淋巴瘤#
18