
早发性难治性癫痫:警惕CDKL5缺乏症(CDD)
2024-12-01 神经科学论坛 神经科学论坛 发表于陕西省

本文介绍发育性癫痫性脑病(DEE)中常见的 CDKL5 缺陷型脑病(CDD)相关内容,包括其发病机制、临床特点、诊断方法、治疗药物及中国专家共识建议等,为了解和应对该疾病提供全面信息。
论坛导读:发育性癫痫性脑病(developmental and epileptic encephalopathies,DEE)是较常见的以幼儿期出现癫痫样发作以及伴有认知功能损伤为特征的一组疾病。目前已有超过90种DEE相关的易感基因被发现,这其中由于丝氨酸-苏氨酸激酶细胞周期蛋白依赖性激酶样蛋白-5(cyclin dependent kinase like-5,CDKL5)功能缺失型突变引起的CDKL5缺陷型脑病(CDKL5 deficiency disorder,CDD)是最常见的DEE类型。CDKL5综合征是由于CDKL5基因突变所引起的一种罕见的神经发育性疾病,病人主要表现为早发癫痫,以及神经系统发育迟缓导致的认知,运动,语言和视觉功能障碍。CDKL5缺乏症在新生儿中的发病率为约1:40000至1:60000,且患儿多为女性。

2003年,Dr. Kalscheuer在婴儿痉挛症和智力障碍病人中发现染色体易位导致CDKL5蛋白异常表达,首次报导了2例CDKL5基因突变病人的临床症状。此后CDKL5各种突变类型导致疾病的病例陆续被报道,CDKL5基因突变涵盖的表型谱广泛,临床症状与婴儿痉挛症、Rett 综合征、天使综合征、自闭症等存在交叉部分。目前由于CDKL5突变所致的神经发育性疾病统称为CDKL5综合征。据世界卫生组织数据显示,全球约有7‰的人患癫痫,中国约有1000多万癫痫患者。2岁以下儿童发病率约为70/10万,其中遗传性癫痫约占30%。据估计,CDD的发病率为4-6万分之一,其中女孩约占总病例的90%。
CDKL5为类细胞周期蛋白依赖性蛋白激酶5基因的简称,定位于Xp22.13,基因组坐标为ChrX:18,425,604-18,653,628,包含21个外显子,编码1030个氨基酸,编码的蛋白广泛分布在所有组织中,脑,胸腺和睾丸中表达水平最高,在神经元细胞内CDKL5主要位于细胞核中,参与树突极化、轴突生长、脊柱形成,是大脑发育的必须蛋白。CDKL5蛋白在大脑中行使激酶功能,它可以通过磷酸化下游底物,从而改变其他蛋白的活性。有研究者对CDKL5敲除的细胞和动物进行生化研究发现钙离子通道亚型Cav2.3的磷酸化程度显著降低;进一步发现CDKL5敲除后会影响Cav2.3的通道功能,表现为钙离子电流的密度显著上调,表明Cav2.3功能增强;最后通过整体行为学实验,发现CDKL5功能缺陷小鼠会呈现出性别差异性的运动功能及认知功能受损,并且雌性小鼠还会出现癫痫易感性的增强。该研究为CDKL5相关的DEE发病机制提供了新的视角和重要的实验依据。
CDKL5综合征是由于CDKL5基因突变导致的一系列神经系统症状,包括癫痫发作、发育迟滞、自闭症、肌张力低下、厌食症、脊柱侧凸畸形、喂养困难、手失用、语言落后等临床表现。近十年来CDKL5常被人们同Rett综合征、癫痫脑病联系在一起。CDKL5综合征其特征是早发性(通常在出生后2个月内)癫痫发作,通常对多种药物治疗无效。CDKL5综合征患者的发育严重受损,只有四分之一的女孩和更小比例的男孩实现独立行走;然而,存在临床可变性,这可能是由基因决定的。胃肠、睡眠和肌肉骨骼问题在CDKL5综合征很常见,就像在其他发育性癫痫性脑病中一样,但大脑视觉障碍的患病率在CDKL5综合征似乎更高。其中几乎所有的CDKL5综合征患者都有癫痫发作。患儿表现出三个阶段的癫痫发作:
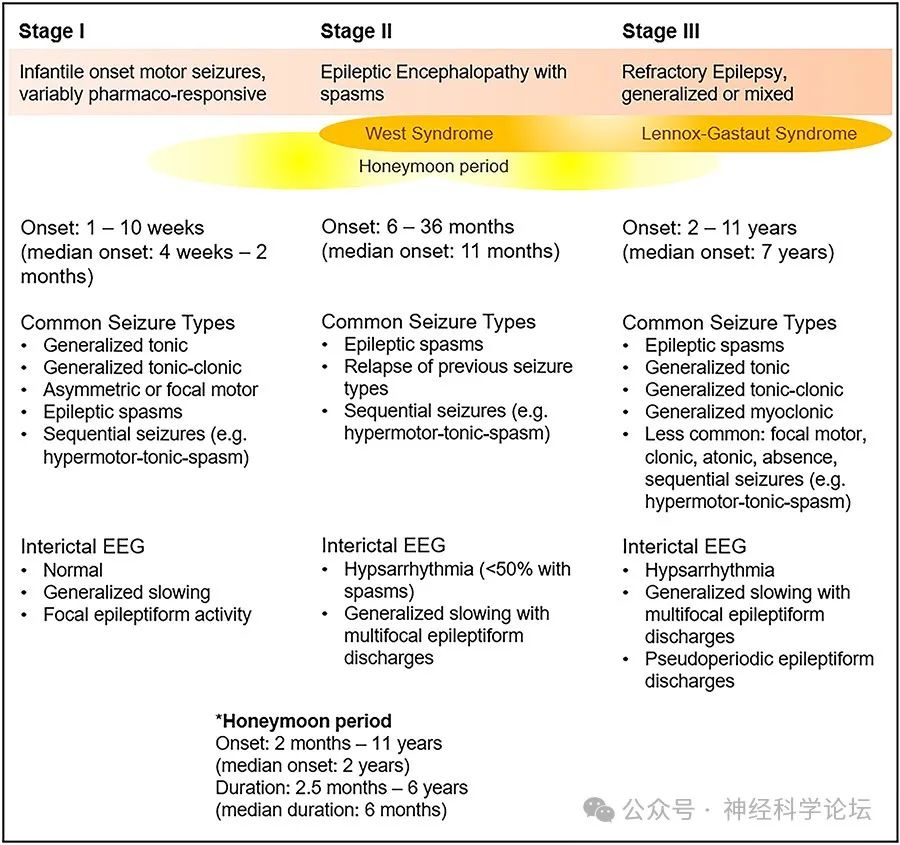
CNS Drugs. 2022 Jun;36(6):591-604.
-
I期是早期癫痫(发病1-10周),脑电图正常。
-
II期是发育型癫痫,通常伴有婴儿痉挛和心律失常,发病率约50%。
-
III期多表现为晚期,多灶性和阵挛性癫痫。
CDKL5综合征临床特点中最主要的特征是早发癫痫。新生儿可能在出生后的数小时,数周或者数月内出现早发癫痫。癫痫发作表现为伴随着肌阵挛的强直发作和强直阵挛性发作,它们通常发生在睡眠中,但随着时间发展,它们也可能会在清醒时出现。有些儿童最初可能被诊断为良性睡眠肌阵挛或Sandifer综合症,还有的儿童会被误诊为Ohtahara综合征,Lennox Gastaut综合征或West综合征,以及Leigh脑病或其他线粒体疾病。但有报告在某些病例中,没有癫痫症状的轻度CDD病可能发生,但与CDKL5基因的特定变异没有明显的相关性。
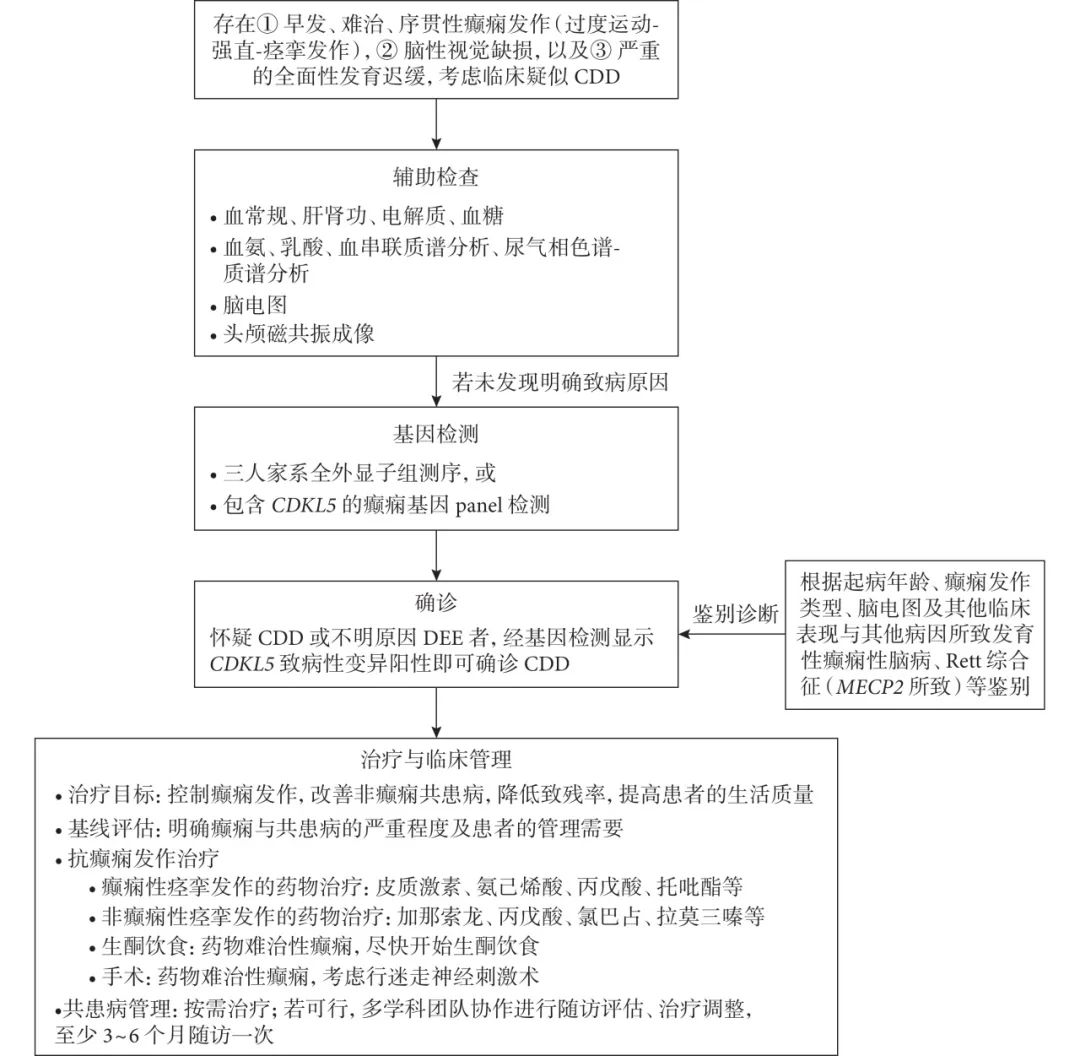
目前通过取病患外周血提取DNA,用基因检测的方式检测是否存在CDKL5突变,可以为该疾病诊断的提供可靠依据。目前CDKL5基因突变所致神经系统疾病均被统称为CDKL5综合征。其遗传学方式为X染色体显性遗传(XD),发病人群主要为女性患儿,但男性患儿不能排除该病。
CDKL5综合征具有难以控制的癫痫发作和重度神经发育障碍等特征。如何控制与之相关的癫痫发作一直是一个难题。2022,美国批准了治疗该病的第一种疗法。该疗法药物目前在欧盟也获得了批准,用于相同适应症。加奈索酮(ganaxolone)是Marinus公司在研的一款靶向GABAA受体的阳性别构调节剂,具有静脉注射和口服两种给药方式。GABA是中枢神经抑制性神经递质之一,与焦虑、紧张、抑郁等情绪变化有关。加奈索酮作用于神经元突触和突触外GABAA受体,从而达到抗癫痫和抗焦虑活性的效用。这是欧盟首个针对儿科患者的治疗方法——Ztalmy(ganaxolone,加奈索酮)口服混悬剂,适用于辅助治疗2至17岁患者与细胞周期蛋白依赖性激酶样5(CDKL5)缺乏症相关的癫痫发作。18岁及以上的患者可以继续Ztalmy。
2023年9月20日, 国家卫生健康委、科学技术部、工业和信息化部、国家药品监督管理局、国家中医药管理局和中央军委后勤保障部等六部门联合发布《第二批罕见病目录》。继2018年国家《第一批罕见病目录》后,本次目录新纳入86种(类)疾病,CDKL5缺乏症被纳入此次罕见病目录。目前,我国两批罕见病目录共收入207种罕见病。在中国,加奈索酮口服混悬剂已于2023年9月8日被CDE以“符合儿童生理特征的儿童用药品新品种、剂型和规格”为由拟纳入优先审评,适用于治疗2岁及以上CDKL5缺乏症患者癫痫发作。
2024年7月16日,元羿生物宣布,其授权引进并商业化的泽元安® (加那索龙口服混悬剂,Ganaxolone)口服混悬剂,已获得国家药品监督管理局批准,用于治疗2岁及以上细胞周期蛋白依赖性激酶5(CDKL5)缺乏症患者癫痫发作。该药物由美国Marinus公司研发,并由元羿生物于2022年授权引进中国。根据协议,Marinus公司将获得1000万美元的预付费,并有资格获得高达2.56亿美元的开发、监管和商业化里程碑付款,以及中国净销售额的两位数分级特许权使用费。

Ztalmy是一种口服小分子神经活性类固醇γ-氨基丁酸A型(GABA-A)受体调节剂。据FDA称,Ztalmy是四氢孕酮的3β-甲基化合成类似物,四氢孕酮是黄体酮的衍生物。在开放标签扩展研究中,接受加奈索酮治疗至少12个月的患者(n=48),主要运动癫痫发作频率中位降低幅度为49.6%。安全性方面,试验中加奈索酮通常耐受良好,并显示与既往临床试验一致的安全性特征,最常见的不良事件为嗜睡。
CDKL5综合征患儿临床表现严重程度与突变位点、突变形式等密切相关,如大多数位于2-12号外显子的突变更为严重,19-21号外显子的变异被认为非致病性或轻度致病性突变。预后与临床表现严重程度密切相关,同时该类患儿需积极抗癫痫治疗同时需细心护理,未规律用药及积极治疗患儿常因癫痫持续状态、肺部感染等疾病早夭,癫痫症状控制后患儿智力发育可出现进步。目前有文献记录的CDKL5综合征患者年纪最大的已经超过了40岁。还有许多患者的年龄在十几岁到三十岁。
2024年10月中国抗癫痫协会创新与转化专业委员会、中华医学会儿科学分会神经学组及中华医学会儿科学分会罕见病学组共同组织领域内专家,结合权威共识推荐、现有研究证据及临床实践经验,制定了《类细胞周期蛋白依赖性蛋白激酶5 缺乏症诊断与治疗的中国专家共识》推荐8条建议:
推荐意见1:CDD 起病早,症状重,表型复杂多样(证据级别2b)。临床应重视CDD的早期识别,及早诊断、及时干预,从而改善患者的预后(专家意见,共识率100%)。
推荐意见2:当患者出现① 早发、难治、序贯性癫痫发作(过度运动-强直-痉挛发作),② 脑性视觉缺损,以及③ 严重的全面性发育迟缓,考虑临床疑似CDD(专家意见,共识率100%)。
推荐意见3:当患者临床疑似CDD,为明确CDD 诊断,建议进行如下基因检测:① 三人核心家系全外显子组测序,或 ② 包含CDKL5基因的癫 痫基因panel 检测 (专家意见,共识率100%)。
推荐意见4:确诊CDD 后,优先使用皮质激素治疗CDD 相关癫痫性痉挛发作(证据等级2b,推荐强度B;共识率97.6%);如皮质激素治疗无效或不耐受,推荐开始使用其他ASMs(包括氨己烯酸、丙戊酸、托吡酯)进行针对癫痫性痉挛发作的治疗(专家意见,共识率100%)。
推荐意见5:在控制CDD 相关的其他癫痫发作时,建议选用加那索龙(证据等级1b,推荐强度A;共识率100%),丙戊酸、氯巴占或拉莫三嗪(证据等级2b,推荐强度B;共识率100%)进行抗癫痫发作治疗。
推荐意见6:针对CDD相关的药物难治性癫痫患者,应尽快开始生酮饮食,作为二线抗癫痫发作治疗的补充(证据等级2b,推荐强度B;共识率100%);如果生酮饮食持续治疗3 个月无效,考虑停用生酮饮食(专家意见,共识率92.7%);如果生酮饮食有效,可持续治疗2年,再酌情逐渐停用(专家意见,共识率97.6%)。
推荐意见7:针对CDD相关的药物难治性癫痫患者,考虑迷走神经刺激术(证据等级2b,推荐强度B;共识率95.1%)。
推荐意见8:CDD 是多系统受累疾病,如果条件允许,建议确诊后由多学科团队协作进行多学科随访评估、治疗调整,且至少3~6个月随访一次(专家意见,共识率100%)。
参考文献
Leonard H, Downs J, Benke TA, Swanson L, Olson H, Demarest S. CDKL5 deficiency disorder: clinical features, diagnosis, and management. Lancet Neurol. 2022 Jun;21(6):563-576. doi: 10.1016/S1474-4422(22)00035-7.
Sampedro-Castañeda M, Baltussen LL, Lopes AT, Qiu Y, Sirvio L, Mihaylov SR, Claxton S, Richardson JC, Lignani G, Ultanir SK. Epilepsy-linked kinase CDKL5 phosphorylates voltage-gated calcium channel Cav2.3, altering inactivation kinetics and neuronal excitability. Nat Commun. 2023 Dec 11;14(1):7830. doi: 10.1038/s41467-023-43475-w.
Aznar-Laín G, Fernández-Mayoralas DM, Caicoya AG, Rocamora R, Pérez-Jurado LA. CDKL5 Deficiency Disorder Without Epilepsy. Pediatr Neurol. 2023 Jul;144:84-89. doi: 10.1016/j.pediatrneurol.2023.04.015.
Hong W, Haviland I, Pestana-Knight E, Weisenberg JL, Demarest S, Marsh ED, Olson HE. CDKL5 Deficiency Disorder-Related Epilepsy: A Review of Current and Emerging Treatment. CNS Drugs. 2022 Jun;36(6):591-604. doi: 10.1007/s40263-022-00921-5.
类细胞周期蛋白依赖性蛋白激酶5缺乏症诊断与治疗的中国专家共识.癫痫杂志,2024,10(6):467-477.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





#CDKL5缺乏症# #发育性癫痫性脑病#
6