
罕见病例|B型尼曼-皮克病及其肝脏受累的异质性表现1例
2024-04-25 临床肝胆病杂志 临床肝胆病杂志 发表于陕西省

现将收治的1例NPD-B病例报道如下,并着重对NPD-B肝脏病变的异质性表现进行文献复习,旨在提高疑难罕见肝脏疾病的临床诊治水平。
尼曼-皮克病(NPD)是一种常染色体隐性遗传的迟发性溶酶体贮积病,本病于1914年首先由Niemann报道,1922年Pick详细描述了病理学检查特征,故而得名,此后国内外陆续有个例报道。A型尼曼-皮克病(NPD-A)和B型尼曼-皮克病(NPD-B)是SMPD1基因致病性变异引起酸性鞘磷脂酶绝对或相关缺乏的等位基因疾病。C型尼曼-皮克病(NPD-C)由NPC1和NPC2基因致病变异引起,会导致胆固醇和糖脂转运障碍。在各型NPD中,NPD-B发病隐匿,病程较迁延,自然病程表现为进展性脾功能亢进、肝功能衰竭或肺功能恶化。NPD-B肝脏受累的异质性较大,临床表现也复杂多样,从轻度肝功能受损、肝肿大、肝硬化到肝衰竭均有报道。现将收治的1例NPD-B病例报道如下,并着重对NPD-B肝脏病变的异质性表现进行文献复习,旨在提高疑难罕见肝脏疾病的临床诊治水平。
1病例资料
女性患者,36岁,浙江金华浦江县人,电商从业者,因“发现肝脾肿大10年”于2020年6月24日入住浙江大学医学院附属邵逸夫医院。患者10年前婚检时发现脾肿大,无任何特殊不适。8年前因“乏力”就诊当地医院,彩超提示:门静脉及脾静脉增宽,门静脉高压;脾肿大。腹部CT:肝脾肿大;门静脉属支曲张,门静脉高压。骨髓穿刺报告提示:造血旺盛,巨噬细胞增多骨髓象。请当地血液科会诊:不考虑原发性血液系统疾病导致肝脾大伴脾功能亢进,有脾切除指征,患者及家属要求暂缓手术出院。在此后的8年间,患者曾多次在多家医院就诊,检查均示血三系减少,肝酶基本正常,凝血功能正常,病因均未能明确。近半年来,患者乏力加重,伴反酸嗳气,偶有右上腹不适感,无恶心呕吐,无呕血黑便,无胸闷气急,无皮肤瘀斑,无肤黄尿黄,为进一步确诊收住本院。门诊拟“肝脾肿大待查”收入。既往史:2003年双侧腋下狐臭手术,2010年剖宫产。来院前2020年5月胃镜检查示:急性糜烂性胃炎;彩超示:门静脉增宽1.5 cm,巨脾,脾静脉增宽。父母及哥哥均体健,儿子体健。长期用药:泮托拉唑、盐酸伊托必利片、康复新液。查体:生命体征平稳,神志清,精神可,全身浅表淋巴结未触及,皮肤黏膜无瘀点瘀斑,巩膜无黄染,无肝掌蜘蛛痣。心脏听诊未闻及杂音,双肺听诊无特殊。腹壁静脉未见,腹平软,无压痛,无反跳痛,肝脏触诊右侧肋缘下4 cm,剑突下5 cm,质地硬如前额,边缘光整,无压痛,无叩痛,脾脏甲乙线6 cm,甲丙线10 cm,丁戊线-1 cm,质地硬如橡胶,边缘光整。墨菲征阴性,麦氏点压痛阴性。肠鸣音听诊正常。双下肢无水肿,病理征阴性。血常规:WBC 2.0×109/L,Hb 101 g/L,PLT 34×109/L。肝功能:ALT 12 U/L,AST 25 U/L,ALP 68 U/L,GGT 21 U/L,Alb 31 g/L, Glb 22 g/L,TBil 8.5 μmol/L, DBil 1.5 μmol/L,ChE 6 980 U/L。凝血功能:PT 12.6 s,INR 0.98。血脂:TC 2.36 mmol/L,TG 3.08 mmol/L,VLDL 0.96 mmol/L,HDL 0.44 mmol/L。感染指标:甲型、乙型、丙型、戊型肝炎血清标志物,EB病毒和巨细胞病毒均阴性。肿瘤指标:AFP、CEA、CA199、CA125均正常。红细胞沉降率10 mm/h,IgG 12.30 g/L,IgA 2.04 g/L,IgM 1.10 g/L,补体C3 0.53 g/L(参考值:0.79~1.52 g/L),补体C4、IgG4均在正常范围,Coombs试验、磷脂抗体均阴性。抗核抗体谱:抗核抗体1∶320颗粒型,SSA抗体阳性,SS-A(RO-52)阳性,SSB抗体阳性。铜蓝蛋白22 mg/dL,铁蛋白正常,转铁蛋白饱和度正常。唾液腺ECT示:左侧颌下腺功能减退,摄锝曲线上升缓慢,酸刺激后泌锝曲线无变化。胃镜检查示:急性糜烂性胃炎,食管和胃底均未见静脉曲张。淋巴结彩超示:全身浅表淋巴结无肿大,边界清,形态规则,皮髓质结构清。心脏彩超示:轻度三尖瓣、二尖瓣反流。肺部及上腹部CT平扫示:两肺未见活动性或间质性病变(图1);肝脾大(图2)。腹部增强CT+门静脉CT血管造影示:肝脾大,其内小脉管瘤考虑;门静脉高压。门静脉主干为主扩张,脾静脉增宽迂曲,脾大,其内多发小斑点低强化灶。肝脏形态正常,体积增大,密度未见明显异常,肝内外胆管未见扩张,胆囊未见异常,胰腺形态无特殊、密度均匀,肾上腺及肾脏未见明显异常,后腹膜未见明显淋巴结肿大,胃壁未见明显异常(图3)。
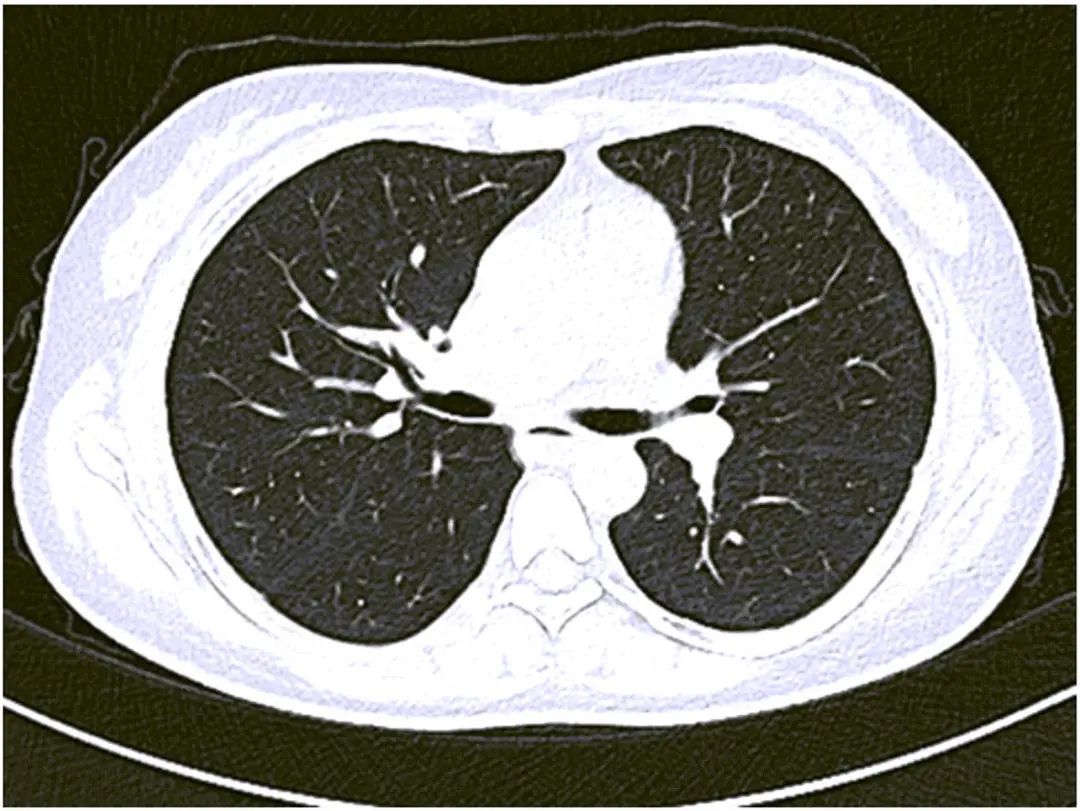
注:两肺未见活动性或间质性病变。
图1 肺部CT
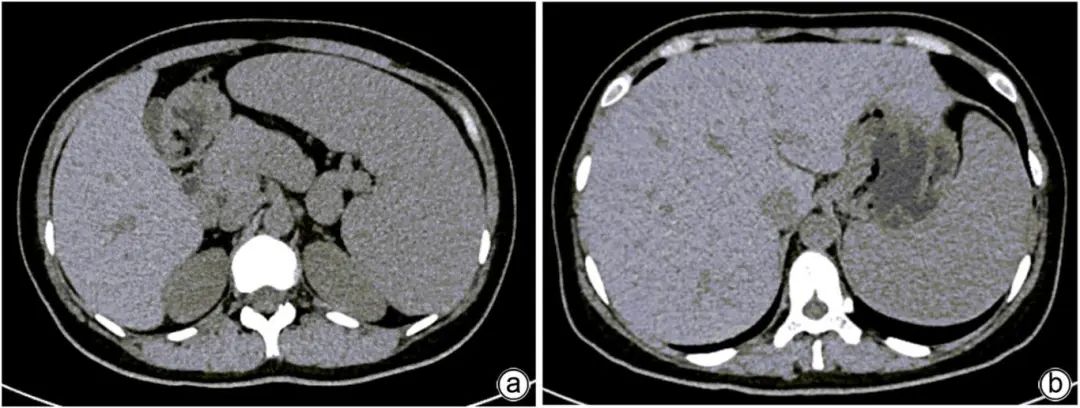
注:a,脾脏肿大;b,肝脏肿大。
图2 上腹部CT平扫
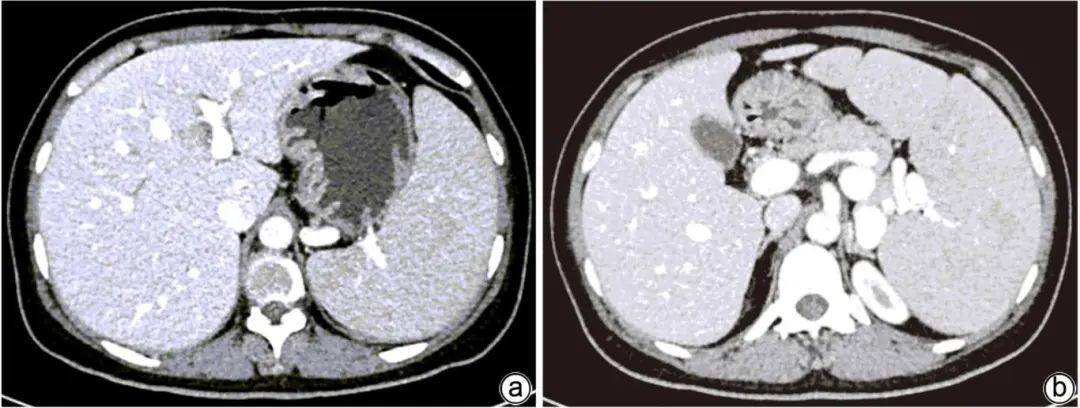
注:a,门静脉扩张,门静脉高压;b,脾静脉增宽迂曲,脾肿大。
图3 腹部增强CT+门静脉CT血管造影
组织病理学检查骨髓常规:组织镜示造血组织约占70%,粒系增生活跃,各阶段粒细胞均可见,以中晚幼及以下阶段细胞增生为主;红系增生活跃,有核红细胞散在分布,幼红细胞簇可见;巨核系增生活跃,数量增多,散在分布,小巨核多见;见多灶组织细胞。骨髓穿刺病理示:骨髓组织增生明显活跃伴红系增生活跃及多灶泡沫细胞(尼曼-皮克细胞),不排除代谢性疾病(图4)。基因检查:对患者进行全外显子高通量测序基因检测,检出基因SMPD1/NM_000543.4转录本1个致病变异和基因SMPD1/NM_000543.4转录本1个疑似致病变异(图5)。对其父母进行了固定位点Sanger测序,具体结果见图6、7。溶酶体酶学及质谱分析:酸性鞘磷脂酶0.91 nmol/17 h.mg(蛋白)[参考值:12.02~114.5 nmol/17 h.mg(蛋白)],壳三糖酶0.76 nmol·mL-1·h-1(血浆)[参考值:3~47 nmol·mL-1·h-1(血浆)]。7-酮胆固醇65.44 ng/mL(参考值:<5 ng/mL)。
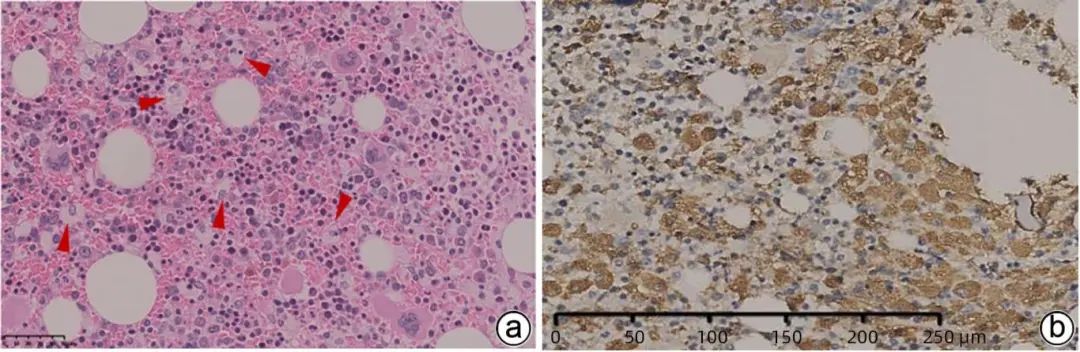
注;a,骨髓组织增生活跃伴多灶泡沫细胞(尼曼-皮克细胞),箭头所示细胞体积大、胞质淡染且含吞噬颗粒(HE染色,×100);b,骨髓组织CD68阳性细胞大量表达,提示为吞噬脂质的尼曼-皮克细胞(DAB染色,×400)。
图4 骨髓穿刺病理组织学检查
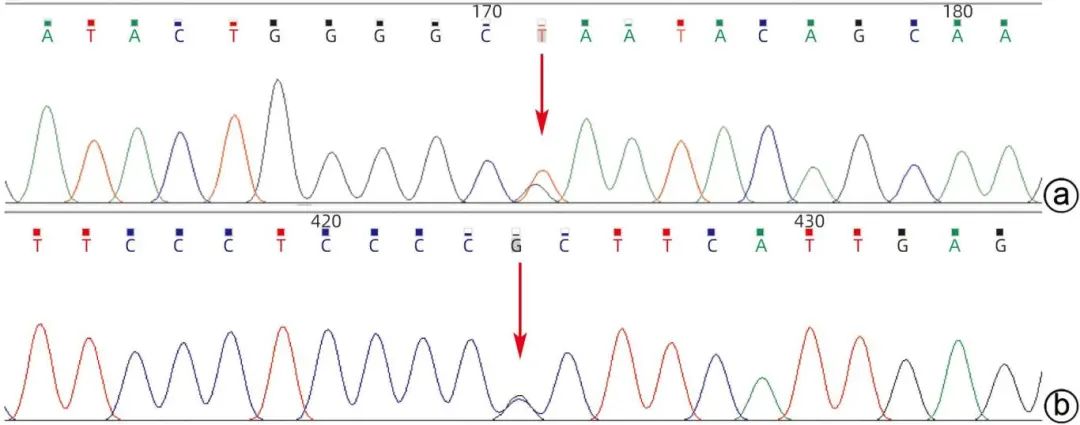
注:a,SMPD1_ex2 c.742G>T(p.Glu248*)受检者Sanger测序验证图(正向,检出),NCBI参考序列ATACTGGGGCGAATACAGCAA;b,SMPD1_ex2 c.995C>G(p.Pro332Arg)受检者Sanger测序验证图(正向,检出),NCBI参考序列TTCCCTCCCCCCTTCATTGAG。
图5 患者遗传病基因检测结果
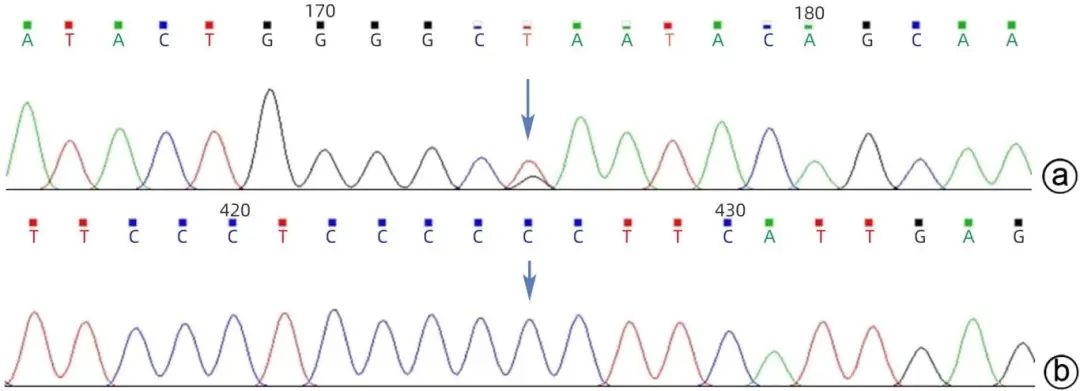
注:a,NM_000543.4(SMPD1)c.742G>T(p.Glu248*)Sanger测序验证图(正向,检出),NCBI参考序列ATACTGGGGCGAATACAGCAA;b,NM_000543.4(SMPD1)c.995C>G(p.Pro332Arg)Sanger测序验证图(正向,未检出),NCBI参考序列TTCCCTCCCCCCTTCATTGAG。
图6 患者母亲固定位点Sanger测序的验证报告
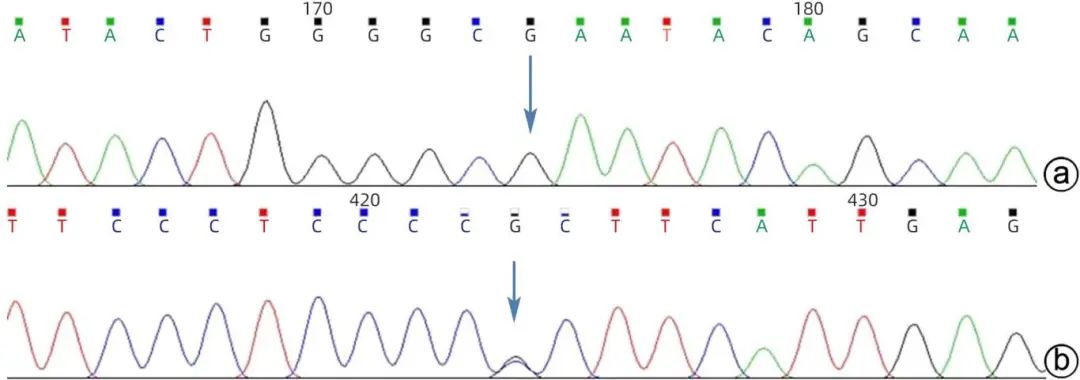
注:a,NM_000543.4(SMPD1)c.742G>T(p.Glu248*)Sanger测序验证图(正向,未检出),NCBI参考序列ATACTGGGGCGAATACAGCAA;b,NM_000543.4(SMPD1)c.995C>G(p.Pro332Arg)Sanger测序验证图(正向,检出),NCBI参考序列TTCCCTCCCCCCTTCATTGAG。
图7 患者父亲固定位点Sanger测序的验证报告
确诊1年后对患者进行了病情的随访,患者吞咽欠顺畅,偶有胸闷感,无呕血,无黑便,无双下肢水肿,能胜任轻体力工作。血常规:WBC 2.5×109/L,Hb 125 g/L,PLT 55×109/L。肝功能:ALT 39 U/L,AST 56 U/L,ALP 122 U/L,GGT 50 U/L,Alb 45 g/L, Glb 33 g/L, TBil 8.6 μmol/L, DBil 3.3 μmol/L,PT 12.1 s, TC 3.34 mmol/L,TG 2.44 mmol/L。肺功能:肺通气功能正常,肺弥散功能轻度降低,气道阻力正常。骨密度正常。胃镜示:慢性非萎缩性胃炎伴胆汁反流。因未能获得酶替代疗法,患者目前服用莫沙必利和氨溴索片对症支持治疗。随访期间,患者病情进展缓慢。
2讨论
NPD-B是一种由编码鞘磷脂磷酸二酯酶-1的SMPD1基因突变导致的常染色体隐性遗传病,发病机制为酸性鞘磷脂酶相对缺乏,不能将鞘磷脂水解为神经酰胺,造成过多的酸性鞘磷脂在患者全身的网状内皮细胞中贮积而致病,临床上以肝脾肿大多见,部分患者伴有肺部浸润,无神经系统症状,大多可活至成人。肝脾肿大是该病大多数患者初诊的原因,肝肿大的程度可达正常时3倍,肝脏触诊质地偏硬,如橡胶状,边缘欠光滑。除了脂质巨噬细胞在脾脏网状内皮系统堆积外,肝脏肿大伴门静脉高压对脾脏肿大有叠加作用,因此,脾脏肿大较肝脏肿大更突出,部分患者脾脏体积可约达正常时28倍,也有脾梗死和脾破裂的报道。本例患者腹部查体发现肝脏和脾脏均肿大,腹部影像学检查提示肝脾大,门静脉、脾静脉增宽等门静脉高压征象,符合NPD-B常见的临床表现。NPD-B患者的肝生化学检查通常为转氨酶轻度升高,以AST升高多见,升高程度多为正常值上限1~2倍。TBil水平也轻度升高,在年长者数值更高。对29例NPD-B患者长达10年随访的一项研究表明,急剧大块的肝细胞坏死是罕见的,随着时间推移,各生化指标大多会保持平稳,仅少数缓慢上升。虽然肝生化学指标大多数为轻度异常,但肝脏病理学改变是多样化的,富含鞘磷脂的泡沫细胞在肝脏中广泛存在,除Kupffer细胞外,还可累及肝细胞、胆管上皮细胞和血管内皮细胞。有报道,中央静脉周围肝细胞肿胀、有细小空泡状细胞质,还可观察到泡沫状组织细胞浸润纤维间隔,而炎症细胞在肝组织中却不常见,部分患者可有轻微纤维化、进展性桥接纤维化,甚至假小叶形成,提示该病可进展为肝硬化。在酸性硝磷脂酶基因敲除的NPD模型小鼠中发现,组织蛋白B过度表达,从而促进肝纤维化,这与临床肝纤维化、肝硬化的表现相一致。有死于肝衰竭患者尸检显示,肝血窦淤滞,被富含脂质Kupffer细胞完全阻滞,可伴机械性溶血的现象。因此,肝生化学指标并不能全面反映NDP-B病情的严重程度,需结合临床症状、脏器功能、病理和影像学检查等全面综合评估。本例患者因脾功能亢进,PLT计数波动在30×109/L~50×109/L,肝穿刺有潜在出血风险,故未行肝穿刺检查。
分子遗传学检测在NPD-B的诊断中具有重要价值。本例患者先通过全外显子高通量基因测序,发现SMPD1_ex2 c.742G>T(p.Glu248*)发生杂合无义突变,该变异会导致第248位氨基酸由谷氨酸变为终止密码子,使编码蛋白序列提前终止,产生截短蛋白或被降解,可能会对蛋白质的结构和功能产生较大影响,经固定位点Sanger测序发现患者母亲为该变异位点的携带者。已有文献报道在多例NPD患者中检出含该变异的复合杂合变异。患者SMPD1基因还检出另一位点杂合错义变异:SMPD1_ex2 c.995C>G(p.Pro332Arg),该变异位点发生在蛋白Metallo-dependent phosphatase-like结构域,经固定位点Sanger测序发现患者父亲为该变异位点的携带者。已有文献报道在NPD患者中检出2例该变异的纯合子和1例含该变异的复合杂合子。此外,患者体内酸性鞘磷脂酶水平显著下降,仅为正常参考值下限的7.5%,从功能学水平进一步证明了相应的诊断。
本病以对症支持治疗为主,必要时可经胃管喂食以确保营养,有症状性肺病者可通过辅助供氧获益。本例患者长期服用莫沙必利促进胃肠道动力,服用氨索溴抗氧化化痰以控制肺部感染。对于高脂血症的患者,降胆固醇药物可以降低肝脏的游离胆固醇,但未证明可以改变临床进程。人重组酸性神经鞘磷脂酶olipudase alfa作为酶替代疗法有望治疗NPD-B,Ⅱ/Ⅲ期临床试验证实了该药物的有效性,但由于该疾病罕见,临床试验推行困难,该药物仍在临床试验阶段。异体造血干细胞移植是治疗本病的根本方法之一,有学者报道移植后能缓解肝脾肿大,肺部间质性病变好转,但神经退行性改变没有改善,故干细胞移植的指征和时机仍有较大争议。近年来,有因严重肝肺功能衰竭的NPD-B患儿行肝移植的报道,发现除了实质脏器的浸润得到改善以外,神经系统受累的患儿认知和运动能力也得到改善,提示肝移植能够抵消肝脏外的表型缺陷,具体机制仍有待进一步研究。针对NPD的基因疗法还处于动物实验阶段,且对神经型NPD无效。
总结本例患者诊断曲折的原因:NPD-B患者虽多年前已有肝脾肿大,因无神经系统受累,进展缓慢,且症状轻微,未引起足够重视。首次骨髓穿刺发现巨噬细胞增多,排除血液系统恶性疾病后,临床医生未与病理医生进一步探讨病因。直至笔者团队接诊发现患者肝细胞损伤不明显,反映肝脏合成功能的指标如白蛋白、凝血功能和胆固醇等无下降,然而门静脉高压表现突出,诊断思维从肝硬化性门静脉高压向非肝硬化性门静脉高压转变,从遗传病角度分析不明原因肝病的诊断,进而完善基因检测,并最终在功能学水平上确诊本病。随访1年发现,患者病情进展缓慢,消化道症状略加重,肝功能和肺功能稳定。对于脾脏肿大突出的NPD-B患者,因脾功能亢进存在出血、感染、接触性脾破裂的危害,外院医生曾建议切脾,患者拒绝有创操作选择保守治疗。有关NPD-B的专家共识不推荐切全脾,因切脾后会加重其他脏器鞘磷脂贮积的负担,反而增加肝衰竭和肺功能恶化的风险,当严重脾功能亢进或门静脉高压不可控时,推荐部分脾切除或脾栓塞。本例患者未出现门静脉高压严重并发症,建议避免身体接触性运动,定期随访。对于确诊本病的患者应对其及亲属开展遗传生殖咨询,可以降低其后代患病的风险。
全文下载
https://www.lcgdbzz.org/cn/article/doi/10.12449/JCH240221

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





#尼曼-皮克病# #肝脏受累#
8