
临床研究|肝豆状核变性不同基因型患者的肝病表型及临床特征
7小时前 临床肝胆病杂志 临床肝胆病杂志 发表于陕西省

关于WD患者基因型和肝病表型的相关性还有待探索,本研究旨在对两者的潜在联系进行探究。
肝豆状核变性(WD)是一种由ATP7B基因突变导致的常染色体隐性遗传铜代谢相关疾病。多数WD患者表现为铜蓝蛋白水平下降和肝功能障碍,包括慢性肝病、失代偿期肝硬化甚至是暴发性肝衰竭,而终末期肝病是WD患者的主要死因之一,因此探索影响WD患者肝病表型的因素具有一定的临床意义。
既往研究认为WD患者的基因型是影响其肝病表型的重要因素,尤其是在中国WD患者中常见的c.2333G>T/p.R778L突变(R778L突变)和c.2975C>T/p.P992L突变(P992L突变)。有研究认为携带R778L突变的患者更易表现为肝病表型,并且发病更早,铜蓝蛋白水平更低。Zhang等通过大型WD队列研究,发现P992L突变与WD临床表型无关。对于通常会造成蛋白功能严重损害的截断突变,Okada等和Merle等认为其与极低的铜蓝蛋白水平以及早期肝衰竭相关。然而,Ferenci等研究显示,相比于基因型,性别和年龄对WD患者的表型影响更大。因此,关于WD患者基因型和肝病表型的相关性还有待探索,本研究旨在对两者的潜在联系进行探究。
1资料与方法
1.1 研究对象
选取2008年8月—2023年6月在解放军总医院第五医学中心就诊,具有临床症状并接受基因检测的WD患者。WD的诊断标准基于莱比锡(Leipzig)评分系统,所有纳入患者Leipzig评分均≥4,并排除了其他疾病的存在。从病历中提取以下资料用于分析。(1)一般情况:性别和发病年龄;(2)临床表型:肝脏或神经系统症状,以及角膜K-F环检查情况;(3)实验室检查:肝功能、血常规、凝血常规和铜生化;(4)病理学检查结果;(5)影像学检查结果;(6)基因检测结果。
1.2 研究方法
1.2.1 表型定义和患者分组
根据Ferenci等的分类方法,将肝病表型按照严重程度分为暴发性WD(发病时表现为急性肝衰竭)、失代偿期肝硬化、慢性肝病(慢性肝炎和代偿期肝硬化)。根据ATP7B基因突变情况将患者分为R778L突变组和非R778L突变组;P992L突变组和非P992L突变组;截断突变组和非截断突变组。截断突变包括无义突变和移码突变。
1.2.2 遗传分析
通过Sanger测序和/或二代测序检测患者ATP7B基因。收集患者及其父母静脉血5 mL,用试剂盒提取血液DNA,操作步骤严格按照试剂盒说明书进行。2014年以前入院的患者,采用Sanger测序对ATP7B基因进行检测。使用Primer3 Plus软件设计测序引物,通过PCR进行扩增。纯化后的PCR产物使用ABI 3730XL DNA Analyzer(Applied Biosystems,美国)进行测序。2014年之后入院的患者进行全外显子测序:对取得的DNA样本进行超声打断,采用KAPA Library Preparation Kit(Roche,美国)试剂构建DNA文库,经探针捕获后的DNA样本在Illumina NovaSeq 6000(Illumina,美国)平台进行高通量测序。测序数据使用Burrows-Wheeler Aligner(BWAv0.7.10)与人类参考基因组(GRCp7/hg19)进行比对。高通量测序鉴定出的候选ATP7B变异经PCR扩增后在ABI 3730XL DNA Analyzer(Applied Biosystems,美国)进行Sanger测序验证。
2结果
2.1 一般资料
共纳入WD患者163例,男94例,女69例。中位入院年龄9岁,其中≤18岁者139例(85.28%)。中位发病年龄8岁,从发病到确诊的时间为0~19年。有明确家族史者11例,疑似家族史者6例。
2.2 临床表现
(1)肝脏受损情况:乏力、纳差42例(25.77%),腹痛、腹胀36例(22.09%),皮肤、巩膜黄染28例(17.18%),双下肢水肿18例(11.04%)。ALT或AST升高138例(84.66%),低蛋白血症47例(28.83%)。临床诊断为慢性肝病121例(慢性肝炎82例,代偿期肝硬化39例),失代偿期肝硬化36例(肝硬化合并食管胃底静脉曲张出血15例,肝硬化合并肝性脑病5例),暴发性WD 6例。(2)神经系统受损情况:四肢震颤5例,言语不清、语速缓慢2例,动作迟缓、走路姿势异常2例。(3)角膜K-F环:153例WD患者进行了眼科裂隙灯检查,角膜K-F环阳性59例(38.56%)。(4)铜生化:血清铜蓝蛋白水平降低154例(94.48%);141例患者进行了24 h尿铜水平检测,其中102例(72.34%)24 h尿铜升高。163例WD患者的具体临床特征分布见表1。
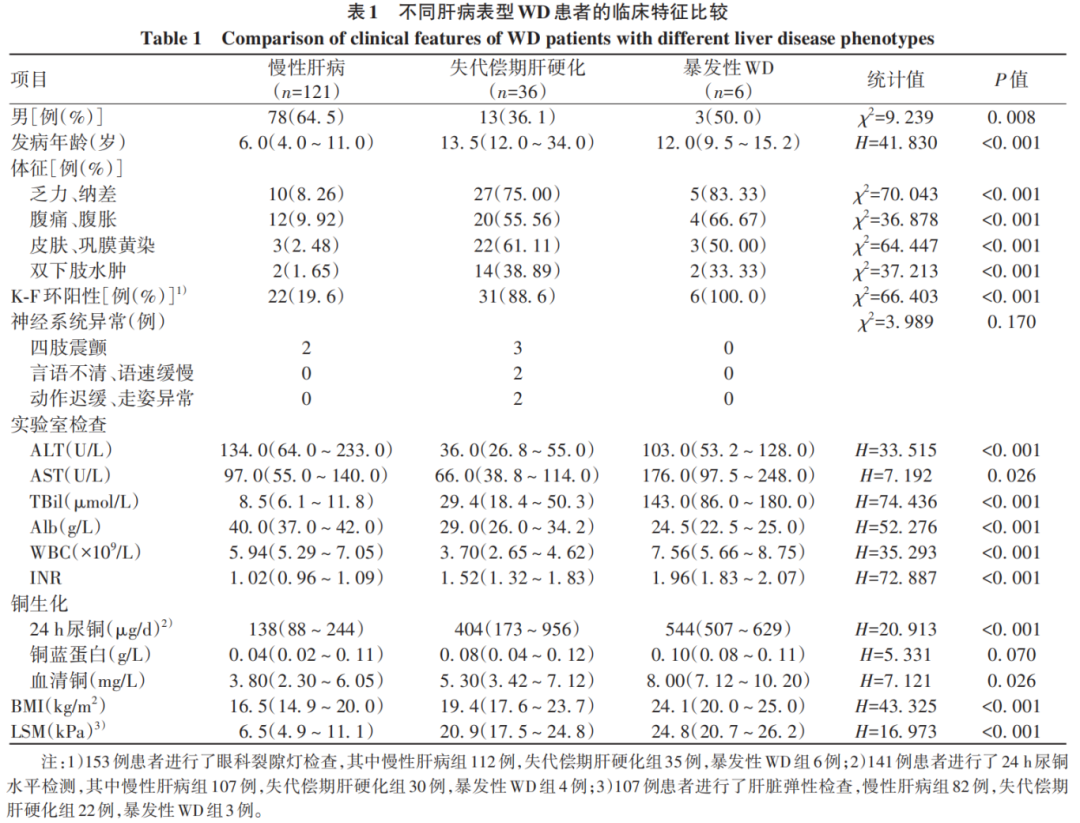
2.3 ATP基因分析结果
163例患者中纯合子21例,复合杂合子137例,杂合子5例。共检测出90种不同的ATP7B等位基因突变,包括61种错义突变、16种截断突变(11种移码突变/5种无义突变)、10种剪切突变、2种外显子缺失和1种同义突变。如图1所示,外显子8的R778L突变是本研究中最常见的突变(等位基因频率为28.2%),其次是外显子13的P992L突变(等位基因频率为12.6%)。截断突变的等位基因频率为11.0%。163例患者的ATP7B基因突变情况见表2。
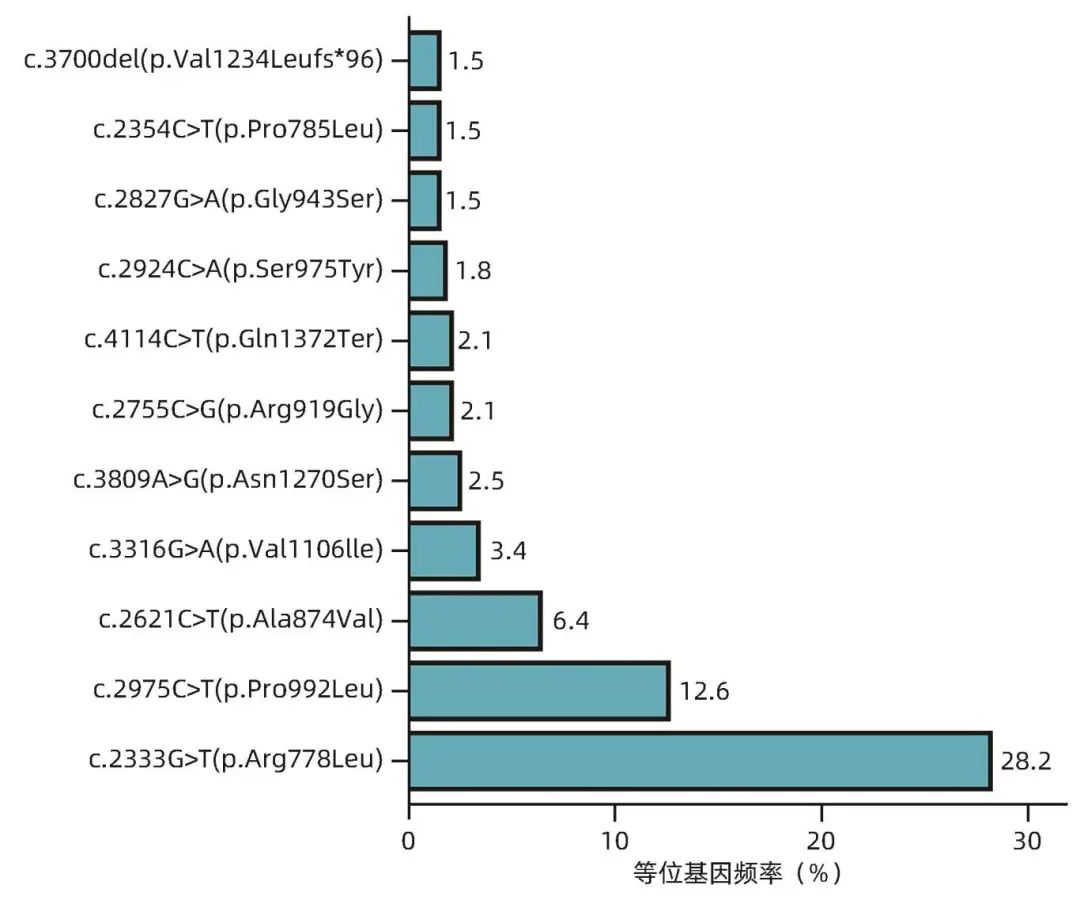
图1 ATP7B基因突变等位基因频率分布图

2.4 R778L突变、P992L突变、截断突变在不同肝病表型中的分布比较
R778L突变、P992L突变和截断突变在不同肝病表型患者中的分布比较,差异均无统计学意义(P值均>0.05)(表3)。
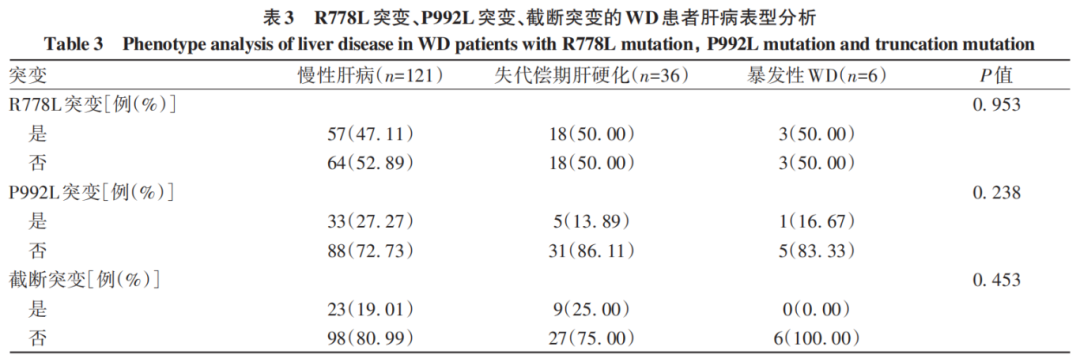
2.5 R778L突变、P992L突变、截断突变的WD患者临床特征分析
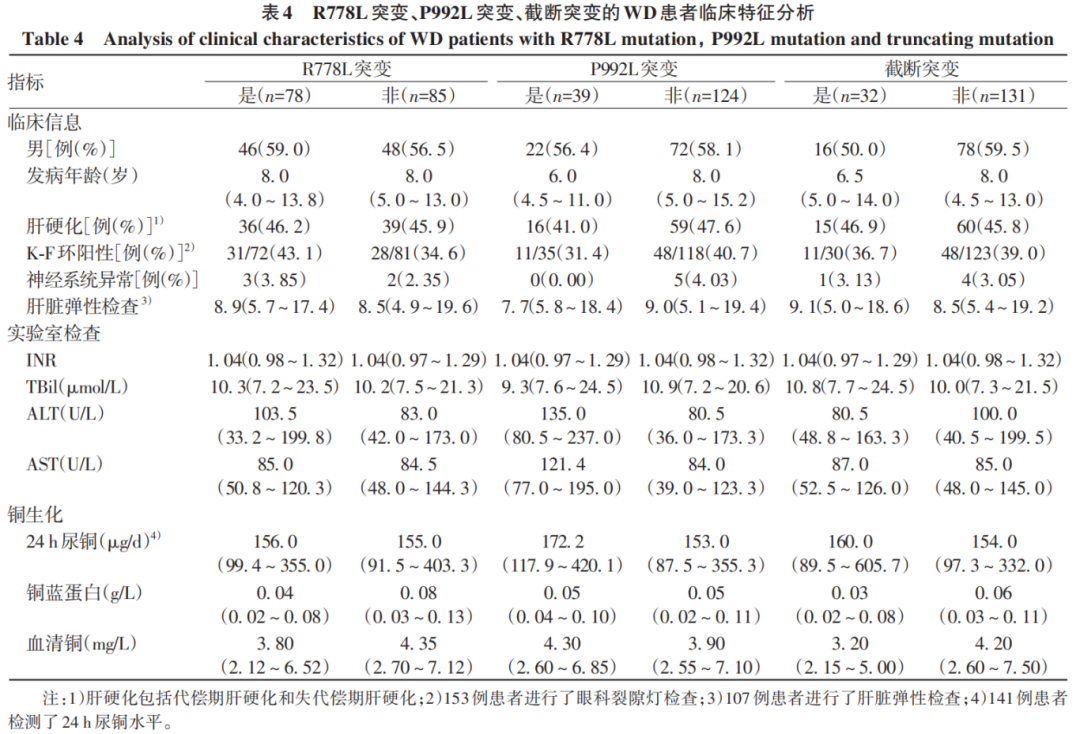
R778L突变组的血清铜蓝蛋白水平显著低于非R778L突变组(Z=-2.889,P=0.004);两组间其他指标比较,差异均无统计学意义(P值均>0.05)。P992L组的ALT(Z=2.684,P=0.007)和AST(Z=3.388,P<0.001)水平均显著高于非P992L突变组;两组间其他指标比较,差异均无统计学意义(P值均>0.05)。截断突变组的血清铜蓝蛋白(Z=-3.136,P=0.002)和血清铜水平(Z=-2.296,P=0.025)明显低于非截断突变组;两组间其他指标比较,差异均无统计学意义(P值均>0.05)(表4)。
3讨论
目前已经发现了超过900种ATP7B基因突变,其中约380种具有致病性。东亚地区最常见的突变为R778L突变,等位基因频率在16.7%~37.9%。本研究中R778L突变等位基因频率为28.2%,与既往研究相符,而欧洲常见的p069Q突变未在本队列中发现。ATP7B基因突变是WD的病理生理基础,突变导致ATP7B蛋白(Cu-ATP酶)活性降低,理论上会影响患者的肝病表型,但目前WD基因型表型的相关性尚不明确。
本研究在163例患者中发现R778L突变与较低的铜蓝蛋白水平有关,该突变导致ATP7B蛋白跨膜区4(TMS4)结构受到严重影响,引起铜离子的排泄障碍,最终使铜蓝蛋白前体无法与铜结合成为铜蓝蛋白而被降解,表现为铜蓝蛋白水平的降低。同样位于TMS编码区的P992L突变主要影响TMS6的结构,然而本研究并未在携带该突变患者中观察到铜蓝蛋白水平下降,但这些患者转氨酶水平较高,提示TMS6对于铜离子运输及铜蓝蛋白合成的影响可能较TMS更小,但TMS6功能受损后对肝细胞损伤更重,其机制有待进一步明确。截断突变通常会造成蛋白结构功能的严重损害,然而未发现其与更严重肝病表型之间的相关性,仅发现携带截断突变的患者表现出更低的铜蓝蛋白和血清铜水平,这种铜生化的损害可能与截断突变体的定位方式存在关联。相对于其他ATP7B突变体多定位于反式高尔基体网络或内质网,截断突变体通常以弥漫性和簇状的形式定位于细胞质内。
本研究显示R778L、P992L、截断突变均与肝病表型无显著关系,表明在探索影响WD患者肝病表型的因素时,不能局限于基因型。Ferenci等认为相对于基因型,性别与年龄对WD患者表型的影响更大。近年来研究者认为除了ATP7B基因突变,WD表型的形成还可能与DNA甲基化异常、组蛋白修饰异常、染色质重塑等表观遗传学因素以及其他非遗传因素(如环境和性别)有关。因此,在评估WD患者肝病表型时,需要综合考虑这些因素。
本研究尚存在一定的局限性:(1)本研究为回顾性研究,存在一定的偏倚;(2)本研究对于肝病表型的探讨仅限于首诊时的情况;(3)本队列中合并神经系统异常的患者例数少,因此未进行基因型与WD患者神经表型相关性的探索;(4)未考虑环境、表观遗传学以及其他非遗传学因素的影响。
综上所述,本研究认为WD患者的基因型和肝病表型无明显相关性,但R778L突变与较低的铜蓝蛋白水平相关,P992L突变与较高的ALT和AST水平相关,截断突变与较低的铜蓝蛋白和血清铜水平相关。
全文下载
https://www.lcgdbzz.org/cn/article/doi/10.12449/JCp40819

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言












#肝豆状核变性# #基因型#
3