
发育倒退,“美人鱼女孩”的秘密
2024-10-23 梅斯罕见新前沿 MedSci原创 发表于上海

悦悦患 Rett 综合征,本文介绍该病症表现、致病原因、分期、诊断及治疗等。
悦悦(化名)本是个可爱的小女孩, 1岁多会走,也开始会有意识叫“爸爸、妈妈”,当家人还沉浸在悦悦成长的喜悦中时,残酷的现实却打破了这份美好。1岁半左右悦悦突然开始变得不爱说话了,走路也变得摇摇晃晃的,智力也比同龄孩子落后,不听指令,目光不对视,还经常双手手背对拍、甩手、流口水,大小便也不能自理。
起初,在当地医院检查没发现明显的异常,医生怀疑是发育迟缓,嘱咐家长带孩子多做康复运动。虽然做了2年的康复训练,但是效果但效果都不明显。于是,悦悦父母带着她四处寻医,来到了广东某医院。
接诊医生发现悦悦对外界反应慢、学习能力差,查体发现患儿发音欠清晰,偶尔能指认自己的五官及日常生活图片,但不能表达感受,不会匹配颜色板、指认动物卡片、理解大小等。患儿的四肢肌力肌张力正常,走路、上下楼梯都正常。结合悦悦的既往病史病症及相关检查报告,悦悦被确诊为:Rett综合征;言语障碍;精神发育迟缓。医生建议悦悦父母为孩子接受“颈动脉外膜剥脱术+迷走神经隔离术。术后,悦悦伤口恢复较好,第三天就能下地走路。接下来她将继续坚持做语言及认知相关康复训练。
美人鱼女孩
Rett综合征(Rett syndrome, RTT)是一种严重影响儿童精神运动发育的神经系统疾病,主要表现为智力低下、语言功能丧失、运动障碍、手部刻板动作、孤独症等。Rett综合征被认为影响女性智力发育的第二大重要原因。Rett综合征发病率大约为1/15000~1/10000,由于Rett综合征主要累及女性,有人将这一群体称为“美人鱼女孩”。当然,也有少数男性Rett综合征的个案报道。
致病原因
MECP2基因是引起Rett综合症最主要的致病基因,不典型Hanefeld型Rett综合征的主要致病基因为MECP2和CDKL5基因,不典型先天型Rett综合征的主要致病基因为MECP2及FOXG1基因。
临床表现
典型Rett综合征的临床特征为:出生后6~18 个月生长发育基本正常,随后出现神经系统发育停滞或倒退,逐渐丧失已获得的技能(如手功能、语言等),出现手的刻板动作(如搓手、绞手、吃手等),步态异常或无法行走,可伴有呼吸异常、便秘、睡眠障碍、惊厥、磨牙、孤独症样行为等,到疾病后期还可能出现骨骼改变,如脊柱侧弯及强直等。
非典型Rett综合征患者临床严重程度不一,或可较典型Rett综合征患者轻,或可较典型者重。根据患者具体的临床表现,非典型Rett综合征可分为语言保留型(Zappella 型)、先天型(Rolando 型)和早发癫痫型(Hanefeld 型)。语言保留型 Rett 综合征患者表现相对较轻,倒退期后可保留部分语言功能和手功能,癫痫、脊柱侧弯、自主神经症状等表现较少见,此型患者生长发育情况一般正常。早发癫痫型Rett综合征以生后早期即出现癫痫发作为临床特征,发作形式多样,药物一般难以控制发作。先天型Rett综合征以生后即出现全面发育落后为临床特征:生后4月内即出现严重小头,生后5月内即可出现发育倒退,严重的精神发育迟滞,此型患者预后较差,大部分患者不能独走。’
分期
I期生前和生后早期(6-18个月)
无明显异常或轻微异常,可表现为运动发育稍落后。
II期1-3岁或4岁
宝宝多生长缓慢,肌张力降低,表现为智力和运动发育停止或倒退,有15%可能会出现惊厥发作。
III期2-10岁
宝宝神经和肌肉功能持续恶化,肌肉明显僵硬,出现典型的刻板行为,多数宝宝有惊厥发作。
IV期10岁以上
表现为运动功能严重倒退,发生肌肉萎缩和骨骼畸形,完全依赖轮椅移动。
Ⅴa:原来有行走功能,但是在这一期丧失;
Ⅴb:从未有行走功能,临床症状更严重。
诊断
Rett综合征诊断标准量表是全世界通用的诊断工具。医生将仔细评估孩子的早期生长发育情况,与当前的体格发育和神经系统发育情况相结合,再给出Rett综合征诊断。发现MECP2突变并不是诊断的必备条件。只要患儿符合以下某种类型。
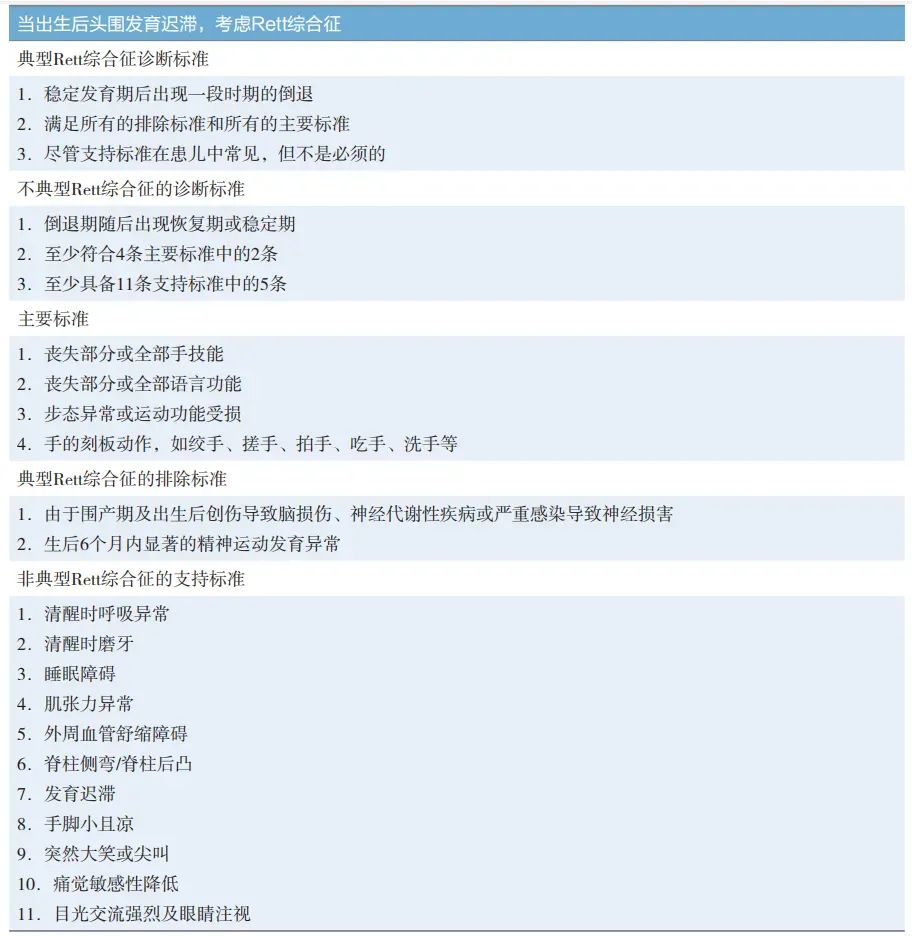
Rett综合征目前无特异性的治疗方法,主要以对症支持治疗为主,如抗癫痫治疗、康复训练、音乐治疗等。对于Rett综合征患者强调的是全生命周期的多学科综合管理,需神经内科、康复科、营养科、消化科、呼吸科、骨科等多学科参与。康复治疗可维持、改善患儿现有的功能,增强运动能力,减缓关节、肌肉变形、挛缩,协调平衡等,减缓其发育倒退进程。
令人兴奋的是,美国FDA于2023年3月10日批准了首个用于治疗Rett综合征的药物DAYBUE(trofinetide,曲芬尼肽)上市,曲芬尼肽可通过减少神经炎症、改善突触功能等发对Rett综合征患者的临床症状(如手部行为、行走和站立等)有不同程度的改善。
参考资料:
[1] Li MR, Pan H, Bao XH, Zhang YZ, Wu XR. MECP2 and CDKL5 gene mutation analysis in Chinese patients with Rett syndrome. J Hum Genet, 2007, 52(1): 38–47.
[2] Neul J L,Fang P,Barrish J,et al. Specific mutations in Methyl-CpG-Binding Protein 2 confer different severity in Rett syndrome.Neurology,2008,70( 16) : 1313-1321.
[3] 何雯洁,代英,钟敏.Rett综合征的诊断和治疗研究进展[J].中华实用儿科临床杂志,2013(06):462-464.
[4] 关荣伟,李秋炎,傅松滨.Rett综合征的临床实践指南[J].中华医学遗传学杂志,2020(03):308-309-310-311-312.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言










#致病基因# #Rett综合征#
21