
病例报告|表现为多系统萎缩小脑型脊髓小脑性共济失调8型1例
2025-01-08 中国神经精神疾病杂志 中国神经精神疾病杂志 发表于陕西省

本报道旨在提示临床医师,SCA8也可表现为自主神经功能障碍、共济失调、脑桥“十字征”等类似多系统萎缩小脑型的特点,临床工作中要避免误诊、漏诊。
摘 要 报道1例表现为多系统萎缩小脑型的脊髓小脑性共济失调8型(spinocerebellar ataxia type 8,SCA8)患者。该患者为57岁男性,病程4年,以头晕、共济失调为首发症状,后出现自主神经功能障碍、快速眼动睡眠障碍等表现。神经系统查体提示自主神经功能障碍、眼球震颤、构音障碍、共济失调,颅脑核磁共振见脑干、小脑对称性萎缩及脑桥“十字征”。基因检测结果显示ATXN8OS的两个等位基因CTA/CTG重复次数异常增多,确诊为SCA8。治疗上予以改善共济失调、自主神经功能障碍等对症治疗后患者反应良好。SCA8为罕见的运动障碍性疾病,临床异质性高。本报道旨在提示临床医师,SCA8也可表现为自主神经功能障碍、共济失调、脑桥“十字征”等类似多系统萎缩小脑型的特点,临床工作中要避免误诊、漏诊。
关键词
脊髓小脑性共济失调8型;多系统萎缩小脑型;ATXN8OS基因;快速眼动睡眠障碍;自主神经功能障碍
脊髓小脑性共济失调8型(spinocerebellar ataxia type 8,SCA8)是由于13号染色体长臂21区的ATXN8OS基因非编码区CTA/CTG异常重复扩增导致的常染色体显性遗传性神经退行性疾病。SCA8发病率低、临床异质性和遗传异质性高,以成年期发病、缓慢进展性共济失调为主要特点[1-2]。表现为自主神经功能障碍、核磁共振显示脑桥“十字征”的SCA8患者尤为罕见[3],极易误诊为多系统萎缩(multiple system atrophy,MSA)小脑型。本文报道1例表现为MSA小脑型的SCA8型患者,并描述其临床特征及转归。
1 临床资料
患者,男,57岁,右利手。因“头晕伴行走不稳4年”于2024年2月就诊。患者4年前无明显诱因下出现头晕,站立或行走时明显,卧位时症状减轻(于当地医院测卧位血压146 mmHg /102 mmHg,立位血压123 mmHg /91 mmHg)。症状缓慢进展并逐渐出现行走不稳、直线行走困难,平地行走或上下楼梯时无跌倒发作,伴有言语不清,偶见饮水呛咳。家属反映患者夜间睡眠常有梦呓、大声喊叫现象。尿频,大便尚正常。无嗅觉减退、肢体麻木、疼痛,无肢体不自主抖动等症状。起病后体质量无明显下降。2022年至今多次就诊于当地医院及复旦大学附属华山医院,诊断为“脊髓小脑性共济失调?多系统萎缩?”,予金刚烷胺、溴吡斯的明、辅酶Q10、帕罗西汀等治疗后头晕症状减轻,但行走不稳、双下肢无力、尿便障碍缓解不明显。患者既往高血压病史多年,平素血压控制可。否认长期吸烟、饮酒、接触毒物史。父母非近亲结婚,有一弟弟48岁、女儿32岁,均未出现共济失调等临床症状(患者父母已过世,弟弟、女儿拒行SCA8基因检测)。
专科检查:意识清醒,定向力、记忆力、语言理解、视空间及执行功能、言语流畅性大致正常。双眼活动自如,水平眼震,构音障碍,言语欠清晰、吐字稍笨拙,咽反射存在。四肢肌力5级,肌张力正常。双手快速轮替完成欠佳,双侧指鼻试验、指指试验稍差。宽基步态,直线行走不能,闭目难立征阳性。感觉检查未见明显异常,腱反射对称,病理征阴性,脑膜刺激征阴性。
辅助检查:颅脑核磁共振平扫见脑干、小脑对称性萎缩,脑桥“十字征”(图1)。SCA1、2、3、6、7、8、10、12、17、36、DRPLA常染色体显性遗传共济失调亚型及FRDA常染色体隐性遗传共济失调的亚型基因检测,结果显示:患者ATXN8OS的两个等位基因(CTA·TAG)n(CTG·CAG)n重复次数分别为28和107次,其中一个等位基因重复次数超出正常范围(正常人该序列的重复次数≤50次),表现出SCA8的致病特征。
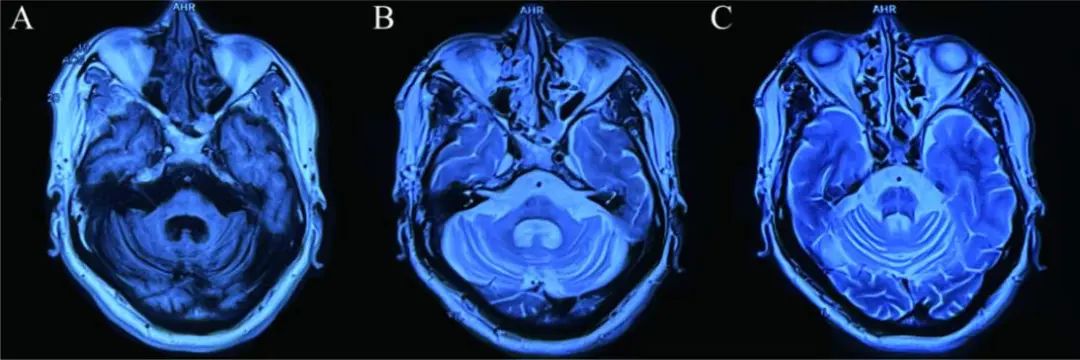
图1 颅脑核磁共振示脑干、小脑对称性萎缩,脑桥“十字征”Fig.1 Brain MR showed atrophy of the brainstem and cerebellum and the “hot cross bun”sign
予多巴丝肼、帕罗西汀、辅酶Q10、溴吡斯的明、维生素B6口服,患者头晕、共济失调稍有减轻,可自行缓慢行走,主观疗效轻度改善。
2 讨论
SCA8于1999年被首次描述,是一组罕见的常染色体显性遗传病[4],起病年龄大多在30~50岁[5]。SCA8的临床特点为成年起病、缓慢加重的小脑性共济失调、构音障碍、眼球震颤。其他表现包括认知障碍、神经精神症状等[4,6-8]。此外,也有少数非典型SCA8特征的患者病例报道,如婴儿期或青少年期发病[9]、皮质基底节变性表型[10]、肌阵挛发作表型[11]、多系统萎缩表型、进行性核上性麻痹表型等[12-14]。SCA8具有较高的临床异质性和遗传异质性,仅凭临床症状来诊断十分困难,确诊依赖于ATXN8OS基因CTA/CTG异常重复扩增序列的检出。
本例53岁起病,初期表现为头晕、直立倾斜试验阳性(血压下降≥20 mmHg/10 mmHg)、共济失调,症状进展缓慢并出现脑干、小脑萎缩、脑桥“十字征”等表现。根据国际MDS运动障碍协会多系统萎缩诊断标准[15]及《多系统萎缩诊断标准中国专家共识(2022)》[16],其符合MSA的核心表现。最初我们推断患者很可能是MSA小脑型。在多系统萎缩小脑型病例中,病程的进展往往预示显著加重的运动障碍和平衡障碍等。但本例随着病程延长,患者运动功能得到较好保留,具体表现为能独立行走及上下楼梯。这使得我们对其MSA小脑型的诊断存疑。值得注意的是,除了MSA外,多种SCA也可能存在自主神经功能障碍[5],多系统萎缩小脑型患者发生眼震和快速眼动睡眠障碍比例为30%~50%[17],SCA患者眼震和快速眼动睡眠障碍的发病率为25%~50%[18-19],二者相仿。且脑桥“十字征”不是MSA的特异性表现,约8.7%的SCA患者也会出现脑桥“十字征”[20-22]。此外,患者虽否认家族史,但其父母去世,且患者弟弟、女儿不同意基因检测,故无法确认其亲属是否为致病基因携带者。为明确诊断,患者进行SCA相关基因检测,证实为MSA表型的SCA8患者,其携带CTA/CTG异常拷贝次数为107次,达到SCA8的诊断标准,诊断明确。分析其父母、女儿未发病可能与SCA8的遗传特征复杂性有关。由于SCA8基因外显率低,不是ATXN8OS基因异常扩增的都会发病。综上,本例SCA8患者临床表现与典型SCA8不同,其合并存在快速眼动障碍、自主神经功能障碍及脑桥“十字征”等表现,既往文献未曾报道。
SCA8发病机制尚不明确,其临床症状、影像学表现的异质性与CTA/CTG基因的扩增数存在相关性[6],与症状的严重程度无显著相关[23-24]。CTA/CTG三核苷酸重复扩增≥80次的患者更容易出现胼胝体发育不良、小脑萎缩、脑干萎缩、第四脑室扩大等影像表现[25-26]。除此之外,不同国家和地区报道的SCA8患者临床表型和CAG/CTG重复次数具有地域特异性,国外健康人群中CTA/CTG重复次数为15~50次,我国人群中致病拷贝数大于70次可辅助临床诊断SCA8亚型[27]。
目前为止,SCA8尚无有效的治疗策略,目前仍推荐以对症治疗为主。临床医生应注意到,“特征性”的影像学特征不是某种疾病的“专属”,SCA8也可以模拟MSA小脑型的症状和影像表现。对于共济失调伴有眼球震颤、快速眼动睡眠障碍等症状体征的患者,即使没有家族史,但是病程进展缓慢时也要考虑到SCA8的可能,需要完善ATXN8OS基因三核苷酸重复次数检测,基因检测对疾病的早期诊断具有重要意义。
参考文献:
1. 陶建霞, 郭硕, 周妍冰, 等. 小脑脊髓性共济失调8型的临床特点及遗传分析[J]. 国际遗传学杂志, 2021, 44(3): 146-151.
2. JUVONEN V, KAIRISTO V, HIETALA M, et al. Calculating predictive values for the large repeat alleles at the SCA8 locus in patients with ataxia[J]. J Med Genet, 2002, 39(12): 935-936.
3. LEE Y C, LIU C S, WU H M, et al. The 'hot cross bun' sign in the patients with spinocerebellar ataxia[J]. Eur J Neurol, 2009, 16(4): 513-516.
4. KOOB M D, MOSELEY M L, SCHUT L J, et al. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8)[J]. Nat Genet, 1999, 21(4): 379-384.
5. CLEARY J D, SUBRAMONY S H, RANUM L. Spinocerebellar Ataxia Type 8[M/OL].1993[2021-04-22] http://www.ncbi.nlm.nih.gov/books/NBK1268.
6. ZEMAN A, STONE J, PORTEOUS M, et al. Spinocerebellar ataxia type 8 in Scotland: genetic and clinical features in seven unrelated cases and a review of published reports[J]. J Neurol Neurosurg Psychiatry, 2004, 75(3): 459-465.
7. TORRENS L, BURNS E, STONE J, et al. Spinocerebellar ataxia type 8 in Scotland: frequency, neurological, neuropsychological and neuropsychiatric findings[J]. Acta Neurol Scand, 2008, 117(1): 41-48.
8. LILJA A, HAMALAINEN P, KAITARANTA E, et al. Cognitive impairment in spinocerebellar ataxia type 8[J]. J Neurol Sci, 2005,237(1-2): 31-38.
9. FELLING R J, BARRON T F. Early onset of ataxia in a child with a pathogenic SCA8 allele[J]. Pediatr Neurol, 2005,33(2): 136-138.
10. BABA Y, UITTI R J, FARRER M J, et al. Sporadic SCA8 mutation resembling corticobasal degeneration[J]. Parkinsonism Relat Disord, 2005, 11(3): 147-150.
11. SWAMINATHAN A. Epilepsy in spinocerebellar ataxia type 8: a case report[J]. J Med Case Rep, 2019, 13(1): 333.
12. SMETCOREN C, WECKHUYSEN D. SCA 8 mimicking MSA-C[J]. Acta Neurol Belg, 2016, 116(2): 221-222.
14. SAMUKAWA M, HIRANO M, SAIGOH K, et al. PSP-Phenotype in SCA8: Case Report and Systemic Review[J]. Cerebellum, 2019,18(1): 76-84.
15. WENNING G K, STANKOVIC I, VIGNATELLI L, et al. The Movement Disorder Society Criteria for the Diagnosis of Multiple System Atrophy[J]. Mov Disord, 2022, 37(6): 1131-1148.
16. 中华医学会神经病学分会帕金森病及运动障碍学组, 中国医师协会神经内科医师分会, 帕金森病及运动障碍学组. 多系统萎缩诊断标准中国专家共识(2022)[J]. 中华神经科杂志, 2023, 56(1): 15-29.
17. STEFANOVA N, BUCKE P, DUERR S, et al. Multiple system atrophy: an update[J]. Lancet Neurol, 2009, 8(12): 1172-1178.
18. PALMA J A, FERNANDEZ-CORDON C, COON E A, et al. Prevalence of REM sleep behavior disorder in multiple system atrophy: a multicenter study and meta-analysis[J]. Clin Auton Res, 2015, 25(1): 69-75.
19. SULLIVAN R, YAU W Y, O'CONNOR E, et al. Spinocerebellar ataxia: an update[J]. J Neurol, 2019, 266(2): 533-544.
20. RIKU Y, WATANABE H, MIMURO M, et al. Non-motor multiple system atrophy associated with sudden death: pathological observations of autonomic nuclei[J]. J Neurol, 2017, 264(11): 2249-2257.
21. HORIMOTO Y, AIBA I, YASUDA T, et al. Longitudinal MRI study of multiple system atrophy - when do the findings appear, and what is the course?[J]. J Neurol, 2002, 249(7): 847-854.
22. WATANABE H, SAITO Y, TERAO S, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients[J]. Brain, 2002, 125(Pt 5): 1070-1083.
23. IKEDA Y, DAUGHTERS R S, RANUM L P. Bidirectional expression of the SCA8 expansion mutation: one mutation, two genes[J]. Cerebellum, 2008, 7(2): 150-158.
24. IKEDA Y, DALTON J C, MOSELEY M L, et al. Spinocerebellar ataxia type 8: molecular genetic comparisons and haplotype analysis of 37 families with ataxia[J]. Am J Hum Genet, 2004,75(1): 3-16.
25. JUVONEN V, HIETALA M, PAIVARINTA M, et al. Clinical and genetic findings in Finnish ataxia patients with the spinocerebellar ataxia 8 repeat expansion[J]. Ann Neurol, 2000,48(3): 354-361.
26. 王丽, 郝莹, 段晓慧, 等. ATXN8OS基因三核苷酸重复扩展突变的脊髓小脑性共济失调患者临床表型特征分析[J]. 中华神经科杂志, 2020, 53(8): 575-581.
27. 王俊岭, 潘乾, 夏昆, 等. 中国汉族人群SCA1、2、3、6、7、8、10、12、17亚型和DRPLA亚型多核苷酸正常重复次数范围研究[J]. 哈尔滨医科大学学报, 2009, 27(5): 501-505.
【引用格式】张亚洁,陈科良,郁金泰. 表现为多系统萎缩小脑型脊髓小脑性共济失调8型1例[J]. 中国神经精神疾病杂志,2024,50(9):557-559.
【Cite this article】ZHANG Y J,CHEN K L,YU J T.A case of spinocerebellar ataxia type 8 presenting with multiple system atrophy cerebral type[J]. Chin J Nervous Mental Dis,2024,50(9):557-559.
DOI:10.3969/j.issn.1002-0152.2024.09.007
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





#多系统萎缩小脑型# #脊髓小脑性共济失调8型#
3