
Sci Bull:哈工大王亚东团队发布迄今最大规模中国人群遗传变异图谱数据资源
2024-06-25 测序中国 测序中国 发表于上海

研究团队从我国13家样本库抽取25,734例中国人样本,进行长-短读长结合的全基因组测序与数据分析,构建了当前最高精度中国人群基因组变异图谱和参考基因组面板。
引言
人群基因组变异图谱是精准医学和个性化医疗的重要科学基础。美欧等数十个国家和地区通过国家计划绘制了本国、本民族基因组变异图谱,推动了重大疾病基因组前沿科学研究与发展。
我国于2018年通过“精准医学研究”重点专项启动了中国人群基因组变异图谱绘制任务。近日,最新成果发表在Science Bulletin。
2024年6月18日,哈尔滨工业大学王亚东教授团队联合国内十余家单位在《Science Bulletin》在线发表题为“A comprehensive genetic variant reference for the Chinese population”的研究成果。研究团队从我国13家样本库抽取25,734例中国人样本,进行长-短读长结合的全基因组测序与数据分析(全部在哈尔滨工业大学生物信息技术研究院完成),构建了当前最高精度中国人群基因组变异图谱和参考基因组面板(Reference panel)。

文章发表在Science Bulletin
研究团队针对现有中国人基因组遗传变异资源存在的人群覆盖不均衡性和缺乏大规模结构变异表征等问题展开研究。通过对25,734例全面覆盖中国七大地理区域的多民族中国人样本进行长-短读长结合的全基因组测序分析(如图1所示),研究团队绘制了当前规模最大的中国人结构变异图谱,其中包含2.79亿个中国人基因组遗传变异(2.54亿个单核苷酸变异(SNV)、2474万个短插入删除变异(Indel)、70万个结构变异(SV)),近1.07亿个新的遗传变异为首次报道(0.97亿个SNV、954万个Indel、13万个SV)。
依据这一图谱:每个中国人平均携带379万个遗传变异,其中包含6462个为未报道过的新变异、276个变异可被注释出与致病性相关。同时,SV变异发现数量也超过前序研究,并发现其对基因组编码区域的影响显著大于SNV和Indel变异。
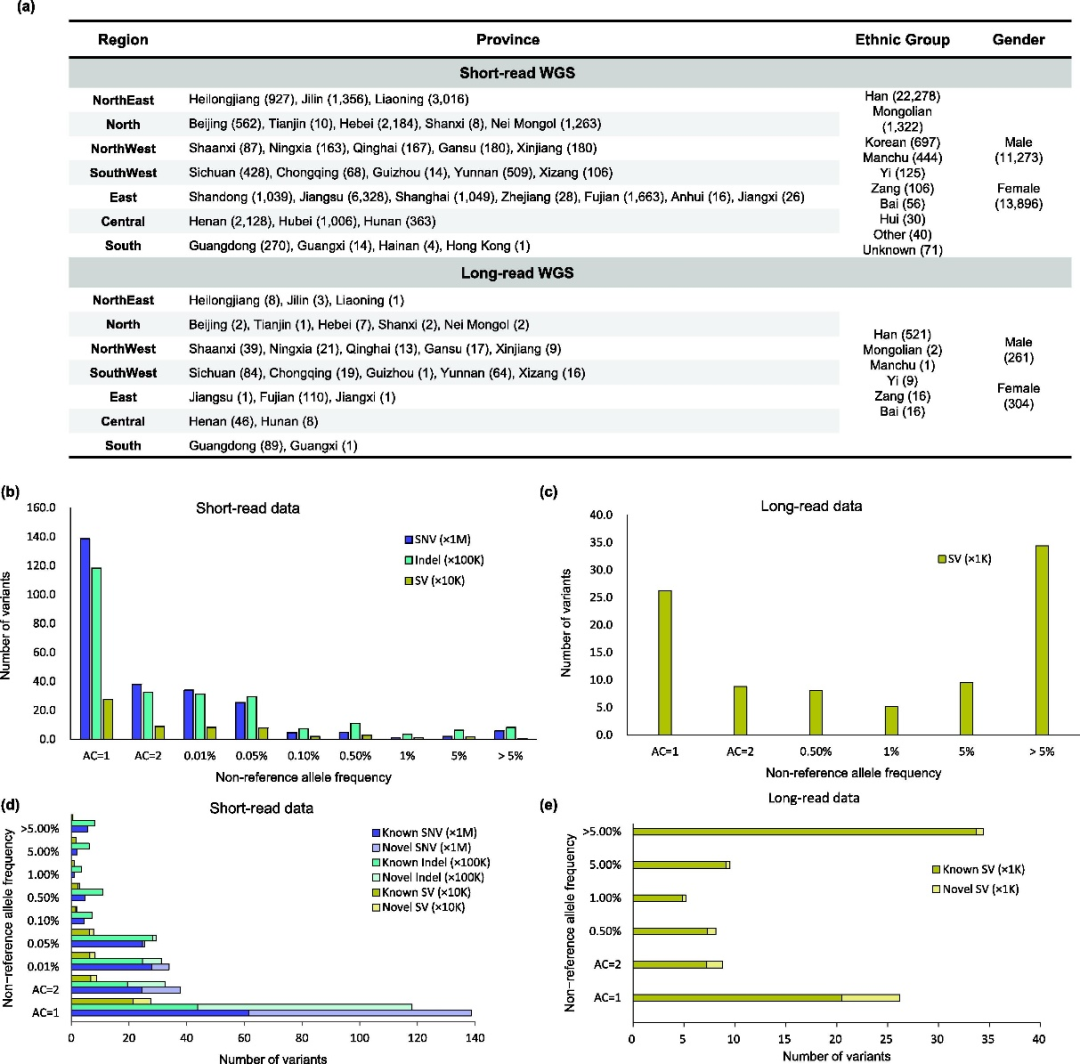
图1. 测序样本与识别变异分布
研究人员还基于此变异图谱构建了参考基因组面板并与国际多个知名的同类型面板进行了比较(如图2所示),结果表明该数据资源可显著提升中国人群变异插补的准确性和敏感性,尤其在低频和罕见变异的插补能力方面表现出色。
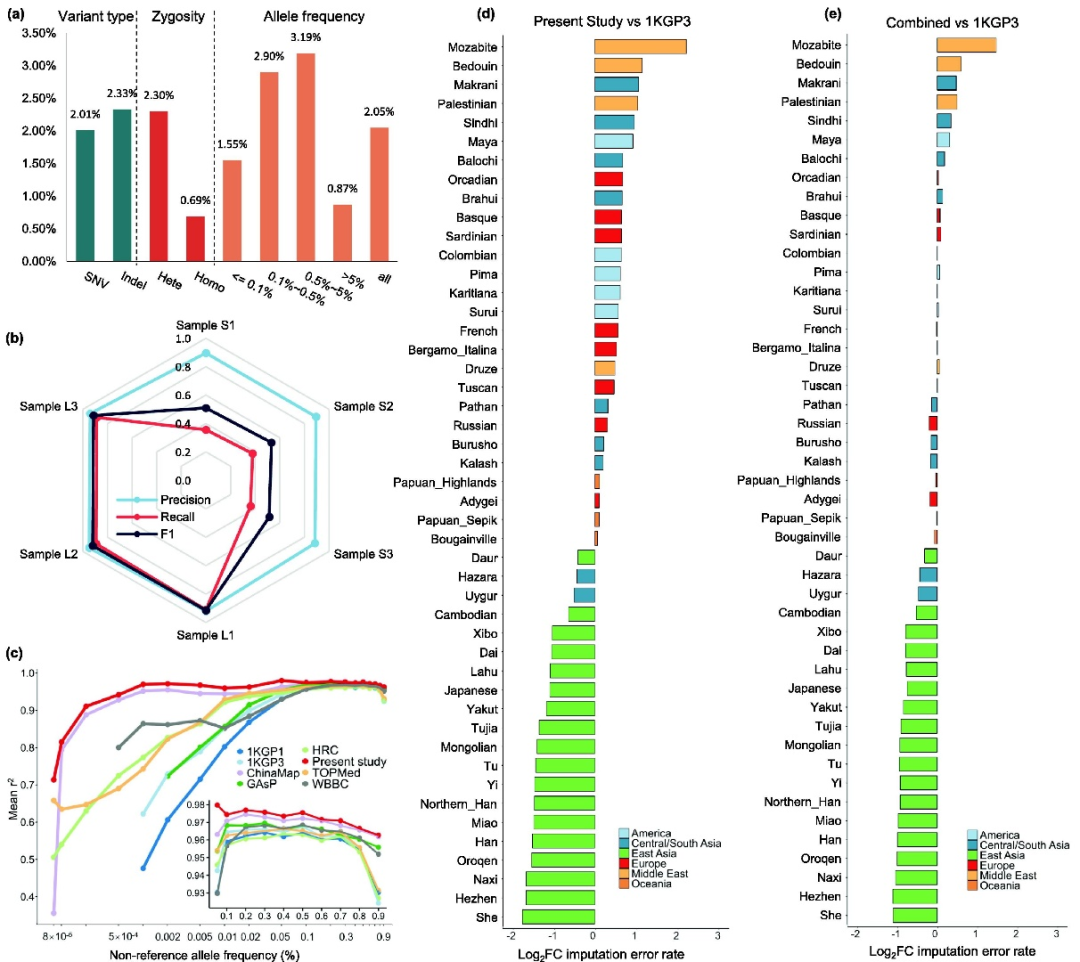
图2. 参考基因组面板性能评估结果
这项研究建立了一个全面的中国人群基因组变异数据资源,并提供了一个可视化的数据库网站平台(http://bioinformatics.hit.edu.cn/chnpop/)、以及一个在线基因型插补服务器(http://bioinformatics.hit.edu.cn/imputation/)。这些资源的推出,旨在为全球研究人员提供便捷的工具,进一步推动人类遗传学研究和相关技术的发展。
结 语
综上所述,该研究构建了当前最大规模的中国人群基因组变异数据资源,全面覆盖SNV、Indel、SV等全谱变异类型。这一遗传数据资源的建立,不仅提供了更广泛的中国遗传多样性特征,还将有力推动我国和世界范围内的精准医学工作的开展,促进人类遗传学等前沿科技领域的发展。
论文原文:
Tao Jiang, Hongzhe Guo, Yadong Liu, et al., A comprehensive genetic variant reference for the Chinese population, Science Bulletin, 2024, ISSN 2095-9273, https://doi.org/10.1016/j.scib.2024.06.017.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#基因组变异图谱# #基因组面板#
43