
慢性光化性皮炎发病机制研究进展
2025-01-01 海龙话皮 海龙话皮

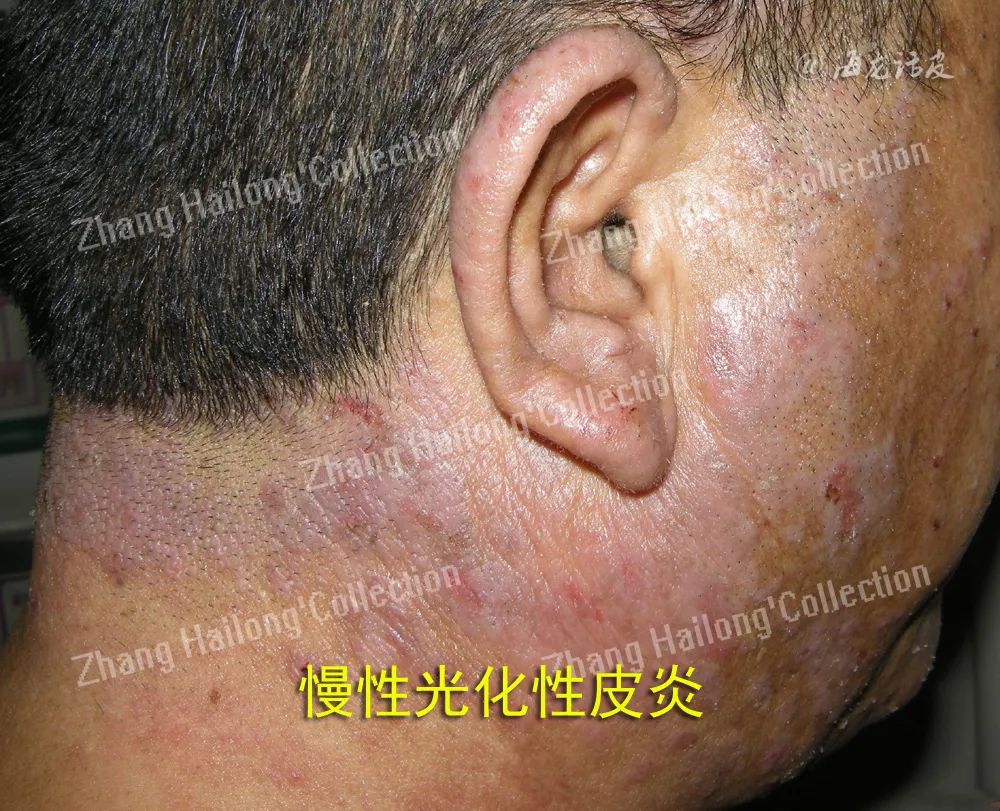
慢性光化性皮炎是慢性、复发性、难治性皮肤病,介绍其临床表现,阐述发病机制,包括紫外线、变应原、免疫应答、氧化应激、细胞凋亡、屏障受损等多方面因素,指出分子水平研究及靶向药研发是趋势。
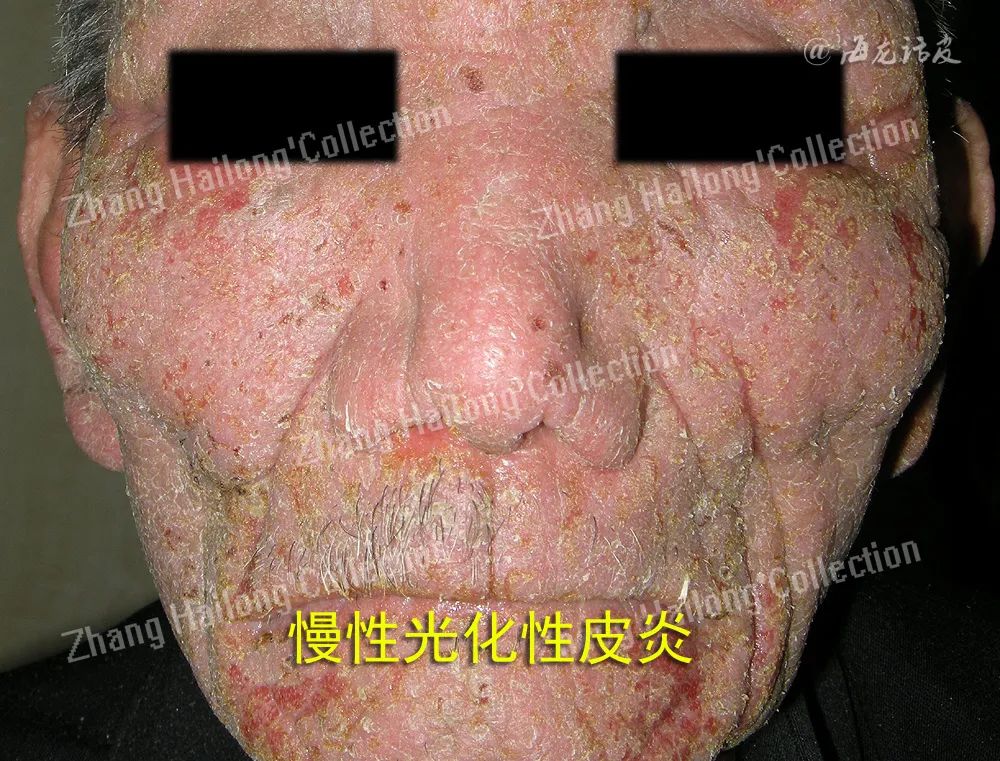
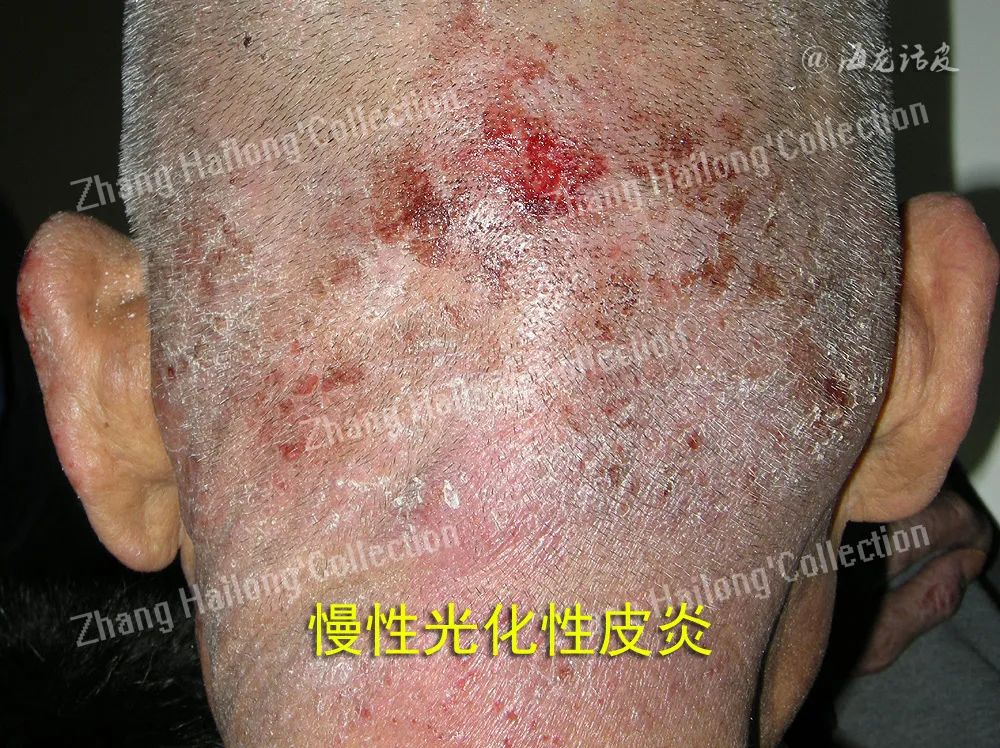
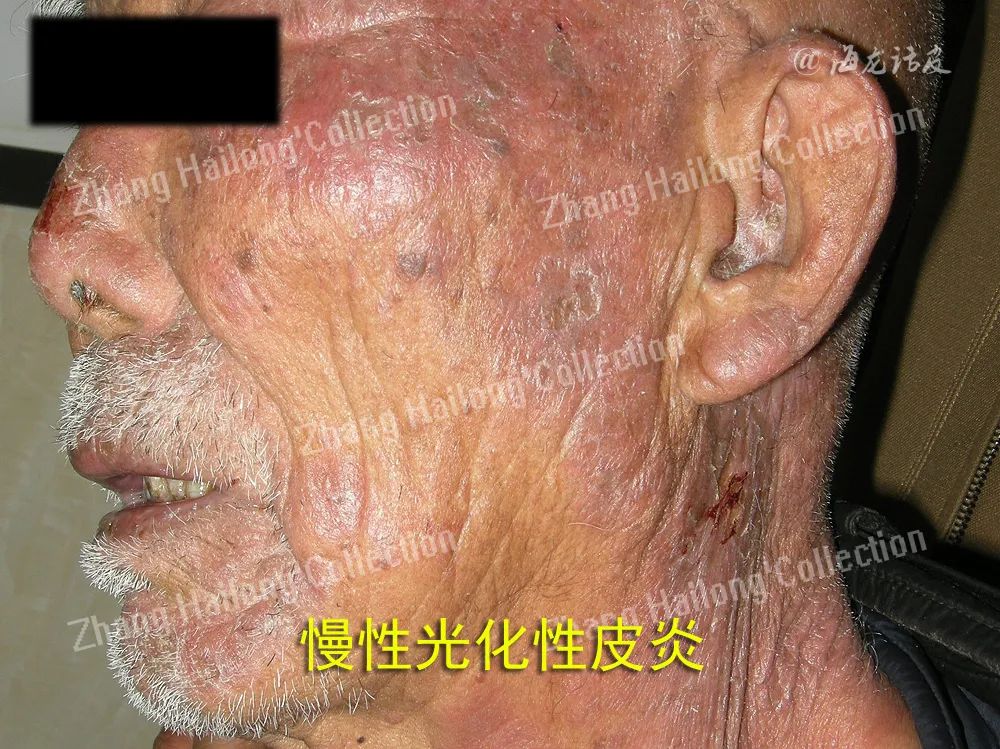
慢性光化性皮炎(chronic actinic dermatitis, CAD)是一种慢性、复发性、难治性光变态接触性皮炎,临床表现为曝光部位,甚至非曝光部位的慢性皮炎湿疹样改变,主要症状为红斑、丘疹、斑块、渗出、结痂、抓痕、皲裂和苔藓样变,伴瘙痒或疼痛,严重者可表现为红皮病。
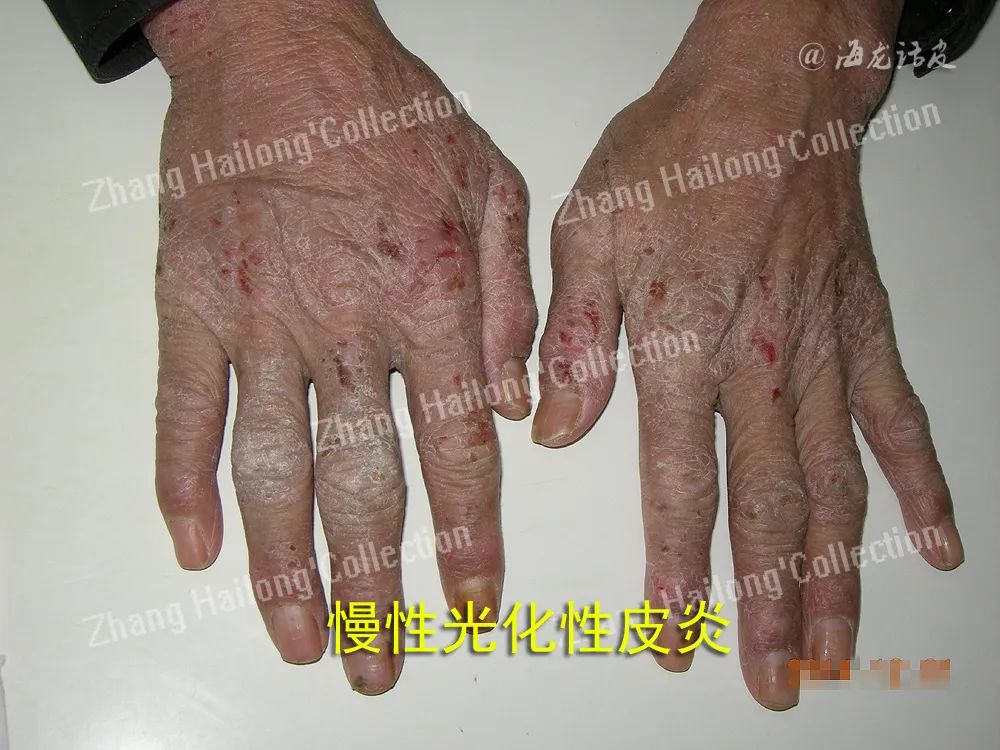
虽然CAD患者临床症状较典型,但其机制极其复杂,目前机制学说如下:患者接触或服用光敏性物质后,曝光部位的皮肤于紫外线或可见光照射后激发了迟发性型超敏反应、氧化应激、细胞凋亡异常、屏障受损是主要机制。此外,感染、特应性体质、性激素紊乱也扮演了重要的角色。
紫外线及可见光直接或间接引起CAD
紫外线是引起CAD的基础。长波紫外线(ultraviolet A,UVA)、中波紫外线(ultraviolet B,UVB)是引起CAD发病的常见原因。轻度患者大部分对UVB敏感,重度患者大部分同时对UVA和UVB同时敏感,少数患者甚至对可见光、短波紫外线(ultraviolet C,UVC)也敏感。不同长度的紫外线到达皮肤的深度不同,UVB到达皮肤的表皮层,而UVA可穿透皮肤真皮层损害细胞DNA引起CAD。
可吸入颗粒物10(particulate matter 10,PM10)与CAD发病率呈负相关,颗粒物多的地方患病率相对较低,可能与紫外线透过减少有关;在污染物、云和雾较少的地方,到达地球表面的紫外线强度较高而引起CAD;而雪、冰、沙子、玻璃和金属可通过反射高达85%的UVB,从而减少紫外线到达地球的辐射量,减少CAD的发生并可能降低疾病严重程度。
接触物和口服药物等变应原引起光变应接触性皮炎
CAD患者长期暴露在紫外线及可见光下,触发一系列变应原引起光变应接触性皮炎。国内外报道CAD患者常见变应原有菊科植物提取物(倍半萜内 酯)、芳香类化合物、金属、橡胶、环氧树脂、三硫化(四)磷、秘鲁香脂、对苯二胺、氯化钴、硫酸镍、药物、防腐剂、载体基质及防晒霜。倍半萜内酯是菊科植物中常见的天然产物,其与胸苷上的环结合形成抗原复合物,引起超敏反应。含有BP-3(氧苯酮)和Bp-4(磺异苯酮)、异丙基二苯甲酰甲烷等成分的防晒霜均可诱发光过敏反应。甚至,农药里面的某些成分可能也是导致光变应接触性皮炎的原因。
Hofmann等总结了包括393种可能具有光敏性的药物,包括神经系统、抗感染类、心血管类、抗炎、抗肿瘤类、内分泌代谢类、其他类型药物,均可能诱发CAD。Blakely等总结了最常见的光敏药物,包括胺碘酮、氯丙嗪、多西环素、氢氯噻嗪、萘啶酸、萘普生、吡罗昔康、四环素、甲硫达嗪、威罗非尼和伏立康唑。
变应原激活迟发性超敏反应并引起免疫应答
异常变应原的有效成分于曝光部位皮肤形成半抗原,与机体载体蛋白结合后形成全抗原,巨噬细胞摄取、加工全抗原并自我激活后引起迟发性超敏反应(delayed type hypersensitivity,DTH)。T细胞接收激活信号后分化为辅助性T细胞CD4+Th和细胞毒性T细胞CD8+Tc,分泌细胞因子引起炎症反应。
CAD患者早期以CD4+Th浸润为主,CD8+Tc细胞随时间延长更占优势,陈旧性皮损以Tc浸润为主,而更多研究表明CAD发生与Tc/Th比例失衡有关,其中,Tc1/Th1在 DTH中占主导作用,而Tc2/Th2占次要作用。Th和Tc细胞可分泌IL-17和IL-22,Treg及Th17细胞是CD4+Th细胞的重要亚群。Treg细胞可维持自身免疫稳定,IL-17可诱导产生IL-6、IL-17、肿瘤坏死因子(TNF-α)致敏,促进IL-17细胞分泌IL-22引起表皮增厚和炎症。
CAD的疾病早期Th17细胞通过分泌细胞因子促进炎症反应,且疾病发展过程中Treg细胞减少可使免疫失衡,Th17/Treg比值与疾病严重程度呈正相关。此外,INF-γ、IL-12增高和CAD有关。Tc1和Th1分泌INF-γ,介导细胞免疫,可活化巨噬细胞和诱导DTH。IL-12诱导静止和活化的T细胞、NK细胞产生INF,同时促进Tc1和Th1分化并抑制Tc2及Th2细胞因子活化。IL-1,IL-6,IL-8可能诱导白细胞分泌而发挥作用。随着时间延长,仅在UVB的照射下,机体载体蛋白的结构就可发生变化而成为内源性抗原,持续刺激免疫系统而引起持续性的迟发性超敏反应。
氧化应激反应与CAP的关系
氧化应激是是指氧自由基引起体内的氧化与抗氧化作用失衡,产生大量氧化中间产物,引起各类疾病。CAD的发生与氧化应激紧密相关。一方面,臭氧是环境中重要的光保护剂,主要吸收UVA和UVC,海拔越高,大气层吸收紫外线的能力越弱,对皮肤细胞DNA的损伤越重,皮肤的氧化应激反应越强。
另一方面,因CAD患者清除超氧化物歧化酶等氧自由基的能力下降,且患者缺乏应对氧自由基损伤的修复能力,从而激发免疫反应。有研究者发现,CAD患者的长链非编码RNAs(long noncoding RNA,lncRNAs)l通过顺式及反式作用影响CAD患者的雌激素通路、Wnt信号通路、皮肤屏障通路、局灶性粘附通路等从而影响氧化应激及免疫反应。
细胞凋亡通路异常表达引起皮肤增厚
CAD与细胞凋亡机制密切相关。皮肤细胞的凋亡和增殖的平衡是维持细胞自身稳定等方面的重要因素。目前更多学者支持CAD与Fas-FasL通路有 关。Fas是一种跨膜蛋白,它与FasL结合可以启动凋亡信号的转导引起细胞凋亡。程浩等认为,虽然FasL在疾病早期高表达,但是Fas的表达随着病程延长逐渐增加,导致皮肤增厚及恶变。凋亡受凋亡抑制因子Bcl-2家族调控,Bcl-2家族的作用是遏制细胞凋亡。
Bcl-2和Bcl-xl是Bcl-2家族蛋白成员,正常情况下,Bcl-xl在角质层最多,而CAD患者Bcl-xl表达增强使表皮增厚明显。hsa-miR-221-3p过表达或FOS抑制可促进细胞增殖并减少细胞凋亡,FOS调节Bcl-xl/bax异二聚体介导hsa-miR-221-3p的角质形成细胞增殖作用。IFI16是干扰素诱导蛋白200家族成员之一,调控细胞增殖与细胞凋亡进程。CAD细胞核内IFI16的减少可下调P53-BAX/BCL-2通路,角质形成细胞周期阻滞和凋亡进程受到影响,并解除了对细胞增殖的抑制,皮损呈增值性改变。
环境和信号通路异常表达造成屏障受损
环境因素可通过影响皮肤屏障从而引起CAD。降水量越少,天气干燥,皮肤角质层水合能力降低,皮肤对UVB抵御能力下降,皮肤屏障易受损。较高的湿度也会通过影响皮肤屏障及改变皮肤结构从而增强UVA的吸收。皮肤老化和晒伤也是皮肤屏障受损的原因。紧密连接是维持皮肤通透功能的结构之一,Occludin是紧密连接的重要组成部分,主要受蛋白激酶C(PKC)的调节。另外,TU等还发现hsa-miR-31-3p的表达在CAD患者中显着上调,引起屏障受损,正调控CSS-CAD和TEWL值。
此外,CAD与HIV或人淋巴细胞病毒I(human t-lymphocyte virus-I,HTLV-I)感染有关。Sidiropoulos等发现部分CAD患者既往存在特应性病史如儿童皮炎,哮喘,过敏或花粉症,说明CAD可能和特应性体质有关,但仍需大量研究证实。
此外,有研究表明性激素紊乱可能参与CAD的发展,是CAD更易发生在男性群体的原因。慢性光化性皮炎发病机制复杂且容易复发,尤其在分子水平研究慢性光化性皮炎,仍有很大探索空间。在分子水平的探索更利于相关靶向药物的研发,控制症状并减少复发的药物研究也是未来的研究趋势。
参考文献:
1.张梅,农祥.慢性光化性皮炎发病机制研究进展[J].皮肤病与性病, 2024, 10(46): 299-305.
2.陈燕 . TNFAIP3和Occludin在紫外线致慢性光化性皮炎发病中的机制研究[D].昆明医科大学,2020.
3.韩洋洋,杨丹琦,王剑锋等.外周血T淋巴细胞亚群及血清可溶性E-选择素在慢性光化性皮炎患者发病中的研究[J]. 中国中西医结合皮肤性病学杂志,2022,21(3):219-221.
3.陈浩,邓丹琪,周晓鸿等.慢性光化性皮炎凋亡蛋白表达的检测[J].临床皮肤科杂志,2005,(8):517-518.
4.其他文献(略)。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言













#期刊论坛#感谢老师分享的研究内容
22
#发病机制# #慢性光化性皮炎#
31