
鼻窦恶性肿瘤分类如何基于分子定义?基因变异对诊断、预后和治疗具有重要意义
2024-04-17 苏州绘真医学 苏州绘真医学 发表于陕西省

本文综述了鼻窦恶性肿瘤的主要分子变异,如DEK、AFF2、NUTM1、IDH1-2和SWI/SNF基因变异,这些基因变异对鼻窦恶性肿瘤的诊断、预后和治疗反应的预测具有重要的实用意义。
近几十年来,头颈部肿瘤的分类不断发展,包括分子检测在鼻窦、唾液腺和好发于头颈部软组织肿瘤中的广泛应用。新分子技术的出现使我们能够定义头颈部独特的多种新型肿瘤类型。此外,针对基因变异的免疫组织化学标志物的不断扩展,有助于快速识别诊断性分子异常。因此,头颈部病理学家目前有可能从分子定义的肿瘤分类中受益,同时仍然主要基于组织病理学和免疫组织化学进行诊断。本文综述了鼻窦恶性肿瘤的主要分子变异,如DEK、AFF2、NUTM1、IDH1-2和SWI/SNF基因变异,这些基因变异对鼻窦恶性肿瘤的诊断、预后和治疗反应的预测具有重要的实用意义。
研究背景
近几十年来,随着分子检测的广泛应用,头颈部肿瘤的分类得到了改进。分子检测不仅可以定义头颈部特有的多种新型肿瘤类型,而且还有助于识别常见的头颈部肿瘤。分子检测已经阐明了已确定但以前难以理解的实体的发病机制,并阐明了各种肿瘤之间的关系。目前的第5版《世界卫生组织(WHO)头颈部肿瘤分类》在很大程度上依赖于分子数据来支持纳入若干新的肿瘤实体及其亚型,并提供详细的预后和发病信息。然而,必须强调的是,在头颈部病理学中,单纯的分子检测不足以做出正确的诊断。事实上,除了现有的组织学形态实体之外,现在分子生物学提供了额外的信息,有助于微调以前不太清楚的亚型和边界。此外,免疫组织化学标志物的范围不断扩大,有助于快速识别有用的诊断性分子特征。因此,头颈部病理学家目前有可能从分子定义的分类中受益,同时仍然主要基于组织病理学和免疫组织化学做出诊断。
近年来,随着染色体易位产生的肿瘤类型特异性融合致癌基因(如DEK::AFF2和NUTM1基因融合)的发现,以及特定肿瘤类型特有的失活抑癌基因的识别,如SWI/SNF缺陷(免疫组织化学可检测到),鼻窦肿瘤分类取得了长足的进展。本文综述了鼻窦恶性肿瘤中对诊断、预后和预测治疗反应有重要意义的主要分子变异。目前,鼻窦恶性肿瘤亟待新的治疗方法。对于大多数组织学亚型,在可行的情况下,手术被认为是治疗的金标准,通常辅以辅助放疗。近年来,采用这种传统方法并没有显著改善患者的生存,目前的研究重点是将现有的治疗策略(如诱导化疗)与之前的创新放疗技术相结合。然而,结果往往仍然令人失望。本文旨在促进对鼻窦肿瘤分子检测和常规诊断方法的交叉作用的认识。
鼻窦上皮性恶性肿瘤
鼻窦道包括鼻腔、鼻旁窦和前颅底,是一个以多种肿瘤为特征的解剖区域,这些肿瘤表现出分子定义实体的显著形态学多样性(表1)。最近发布的第5版WHO头颈部肿瘤分类包括分子遗传学在其中具有重要诊断作用的新实体,包括HPV相关的多表型鼻窦癌。SWI/SNF缺陷型的鼻窦癌和腺癌,以及包括IDH突变恶性肿瘤的一组新兴实体,被分类为鼻窦未分化癌(SNUC)。分子遗传学也在表现为鼻窦遗传性综合征的背景下发挥诊断作用。
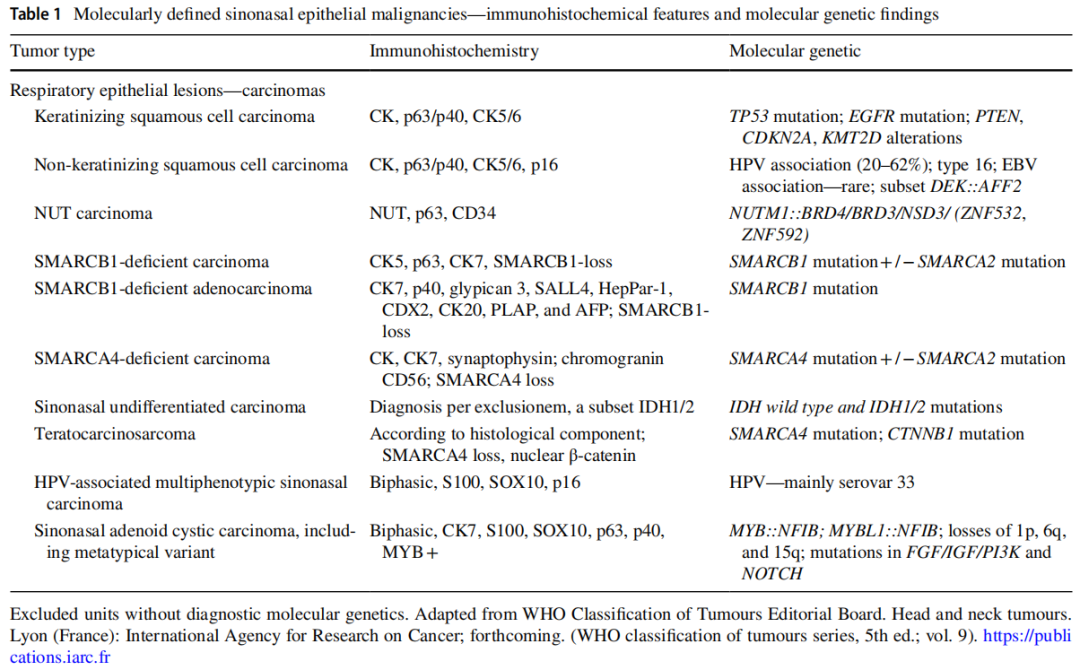
表1
非角化性鳞状细胞癌,包括携带DEK::AFF2基因融合的:
鼻窦非角化性鳞状细胞癌(NKSCC)是一类病因和分子发病机制不同的异质性肿瘤。越来越明显的是,NKSCC作为单纯的描述性诊断已不再有效。值得注意的是,这些肿瘤中有一部分是由具有转录活性的人乳头瘤病毒(HPV)驱动,最常见的是16型,在36-58%的确诊患者中。虽然不建议对鼻窦NKSCC进行常规HPV检测,但偶尔也有助于诊断。由于p16免疫组织化学在鼻窦肿瘤中特异性较差,若要进行HPV检测,必须采用原位杂交或PCR等HPV特异性检测。最近,研究发现超过一半与HPV无关的NKSCC具有频繁易位DEK::AFF2。这些新进展表明,在排除这些基因定义的亚型后,需要对“NKSCC NOS”采用“排除诊断”策略。
DEK::AFF2癌:
DEK::AFF2癌目前被归类为非HPV相关NKSCC类别下的一种新兴实体,尤其局限于鼻窦。不过据报道,也有少数病例发生在中耳、颞骨、眼眶和肺。尽管该肿瘤形态上看起来温和,但其行为方式具有侵袭性,局部复发、转移性淋巴结播散和远处播散的风险高。
组织学上,大多数病例表现出基底样细胞向过渡细胞的复杂的外生和内生生长。如有,乳头状叶的外观从纤细到宽。肿瘤细胞也生长到下面的基质中,形成吻合的小叶、带状,偶有巢状和索状。侵袭方式倾向于广泛的、推挤性的,但也可能表现为非粘连性的侵袭方式,大量不规则的小巢广泛浸润于骨。肿瘤小叶周边常可见核栅栏状结构(图1A)。上皮内细胞分裂可能导致假乳头状形成和肿瘤片中心的星状网状外观。通常,肿瘤细胞具有外观平淡、单调、圆形至椭圆形的细胞核,具有细小至囊泡状染色质、突出的核仁、两亲性至嗜酸性细胞质以及不明显的细胞边界。有丝分裂计数通常较低,但在少数病例中也可见高有丝分裂指数。部分病例可见肿瘤坏死和凋亡小体。大多数肿瘤上皮和间质内均有大量中性粒细胞密集浸润(图1B)。可能会形成微脓肿。
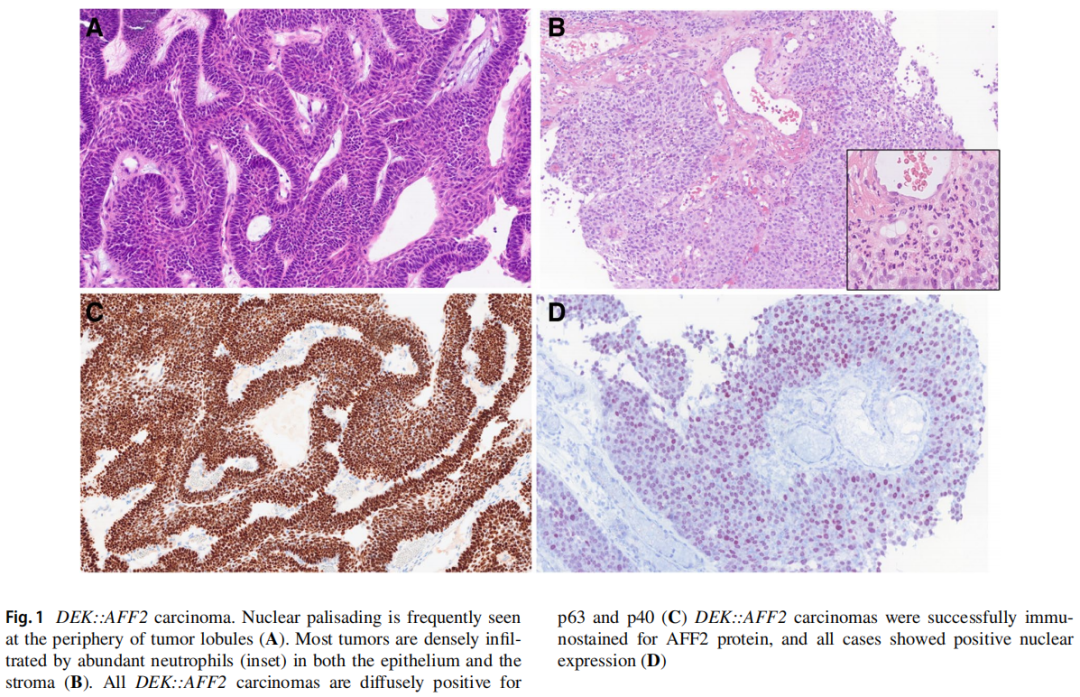
图1
所有DEK::AFF2癌均弥漫阳性表达p63和p40(图1C)。细胞角蛋白AE1/AE3和CK5/6也呈阳性。DEK::AFF2癌可成功进行AFF2蛋白的免疫染色,所有病例均显示阳性的细胞核表达(图1D)。因此,AFF2免疫组织化学是一种新的高度敏感和特异的辅助标记物,可以鉴别DEK::AFF2癌与其他形态特征重叠的鼻窦肿瘤,并可能在脱钙标本中有用。该实体的之前病例报告描述了一例DEK::AFF2癌,对免疫检查点抑制剂(ICI)-抗PD-L1-有很好的反应,这与肿瘤消退期间的DEK::AFF2新抗原特异性T细胞反应有关。
鼻窦NUT癌:
NUT 癌是一种高度侵袭性、大多致命的恶性肿瘤,具有单调的低分化形态。NUT癌(以前称为NUT中线癌)好发于纵隔(约50%的病例)和头颈部,尤其是鼻窦,但也有其他部位,例如喉部。在组织学上,NUT癌是一种分化很差的恶性肿瘤,呈巢状和片状生长。NUT癌是一种高度浸润性和细胞学级别高的恶性肿瘤,有许多核分裂象和常见的肿瘤坏死。诊断的线索是,尽管肿瘤明显为高级别,但肿瘤细胞核缺乏高级别癌中常见的显著多形性。相反,细胞核相对均匀和单调(图2A)。在一些NUT癌中,明显的鳞状细胞分化表现为“突然的”角化,即未分化的肿瘤细胞常紧挨着高分化的角蛋白珠,或者表现为在未分化的基底样细胞聚集物中具有丰富透明细胞质的“突然的”鳞状细胞聚集物(图2B)。这种明显的鳞状分化见于不超过43%的病例。

图2
NUT癌被认为是鳞状细胞癌谱的一部分,细胞角蛋白(尤其是CK5/6,图2C)和p63的阳性支持了这一观点,而p40的阳性不太可靠。NUT单克隆抗体对NUT癌具有高度特异性(图2D)。NUTM1重排肿瘤如皮肤附件孔样肿瘤和CIC::NUTM1肉瘤免疫组织化学染色NUT抗体阳性,但这些肿瘤几乎从不发生在鼻窦。野生型NUT蛋白有限的局灶性表达在一些传统的鳞状细胞癌中很少见,染色较弱且局灶性,出现在< 20%的肿瘤细胞中。典型的点状阳性模式局限于NUT癌,可见于>70%的细胞,并常在所有肿瘤细胞中均呈一致阳性。
NUT癌的基因特征是染色体15q14上NUTM1基因的重排。NUT基因在成熟精原细胞中生理性表达。NUT信号分子与组蛋白乙酰转移酶(HAT)p300结合,激活组蛋白乙酰化。NUT最常见的融合伙伴是BET家族(BRD2、BRD3、BRD4和BRDT)中参与转录和染色体调控的基因。在75%的病例中,NUTM1的融合伴侣是BRD4基因(位于19p13.1),其次是15%病例的BRD3基因(9q34.2)。NUT::BRD3/4融合蛋白通过阻断细胞分化和促进不受控制的细胞生长发挥作用。在一部分病例中,NUT与非BRD基因融合。在6%的病例中,融合涉及NSD3基因(8p11.23),该基因编码分化和细胞增殖调节所需的癌蛋白。在2%的病例中,包含锌指蛋白的基因(18q21.32上的ZNF532或15q25.3上的ZNF592)与NUT融合蛋白有关。
直到最近,对于中位生存期为6.7个月的NUT癌还没有已知的有效治疗方法,这解释了这种侵袭性实体被称为“通往天堂的门票”。手术和放疗是金标准,可延长无进展生存期和总生存期(OS)。最近,诱导化疗策略的结果略好。最近一项研究纳入了MD安德森癌症中心接受治疗的12例鼻窦NUT癌患者,其中位OS为14.6个月。上颌窦肿瘤患者的生存率高出91%(风险比[HR],0.094;95%置信区间[CI],0.011-0.78,p=0.011)。较高疾病分期的患者有较差的OS(IVb-c期无2年生存率,而III-Iva期2年生存率为60%,p=0.05)。所有3例无病生存的患者均接受了诱导化疗。第一个针对NUT癌的靶向药物是组蛋白去乙酰化酶抑制剂(HDACi)(伏立诺他,vorinostat)和BET抑制剂(BETi),它们抑制肿瘤细胞生长并诱导细胞分化。BETi(JQ1)分子模拟乙酰化组蛋白并竞争性抑制BRD3/4与乙酰化染色质的连接。此外,BETi直接靶向NUT融合蛋白。NUT 癌的这种靶向是否会产生临床反应尚未得到研究。此外,迄今为止还没有任何一种BETi获得了FDA的批准。
SWI/SNF复合体缺陷型鼻窦癌及其他恶性肿瘤
SWI/SNF是一种由20多种肿瘤抑制因子组成的多形性复合体,可与启动子位点的转录因子沟通,动员核小体,调节染色质结构。这些基因参与细胞分化和增殖。SWI/SNF复合体缺陷型鼻窦/颅底恶性肿瘤有4种不同的亚型,包括SMARCB1缺陷型鼻窦癌、SMARCB1缺陷型鼻窦腺癌、SMARCA4缺陷型鼻窦癌以及SMARCA4缺陷型畸胎癌肉瘤的一个亚组。低分化脊索瘤也有SMARCB1缺陷,从颅底延伸到鼻窦的情况很少。
SMARCB1缺陷型鼻窦癌:
2014年,两个独立的研究小组首次发现了一个低分化或未分化鼻窦癌亚群中的SMARCB1缺陷,随后又有几个病例系列报道,其中最大的多机构系列报道包括39例患者。SMARCB1缺陷型鼻窦癌的定义为缺乏任何其他定义的鼻窦癌类型的特征、SMARCB1表达完全缺失以及缺乏形态学鳞状或腺体分化。大多数病例表现为基底样细胞形态,但缺乏鳞状特征和角化。然而,约30%的病例显示具有横纹肌样和/或浆细胞样特征的嗜酸性细胞。肿瘤细胞通常呈大的实性巢状和片状。免疫组织化学染色显示,SMARCB1缺陷型鼻窦癌肿瘤细胞均呈全细胞角蛋白阳性,p63/p40呈局灶性阳性,NUT和p16阴性。
SMARCB1缺陷型鼻窦腺癌:
这是一种罕见的SWI/SNF缺陷型恶性肿瘤,定义为存在明确的腺分化和/或存在腺癌的其他特征。肿瘤组织形态学以实性为主,呈小梁状和腺泡状生长。肿瘤细胞较大,呈嗜酸性、嗜酸细胞样、浆细胞样和/或横纹肌样外观(图3A)。SMARCB1缺陷型鼻窦腺癌表现出不同比例的腺体形态,包括伴有流产性微腺分化的肺泡/腺泡结构、小梁结构以及实性/筛状/岛状结构。常可见卵黄囊瘤样分化区域,包括Schiller-Duval体样结构(图3B)。卵黄囊瘤的免疫组织化学标志物(SALL4或磷脂酰肌醇蛋白聚糖-3)常可见(图3C),与卵黄囊瘤样组织学相对应。SMARCB1缺陷型鼻窦腺癌缺乏基底样形态,这在60%-70%的SMARCB1缺陷型鼻窦癌中可见,而SMARCB1缺陷型鼻窦腺癌显示明确的腺体形成、筛状模式或黏蛋白生成。其中一些让人联想到高级别非肠型腺癌,而另一些则可能与肌上皮癌非常相似。卵黄囊瘤样形态仅限于腺癌亚组。虽然可以看到局部p63和/或CK5/6免疫阳性,但缺乏弥漫均匀的鳞状细胞型模式,常表达卵黄囊标志物。然而,具有两种类型之间过渡特征的肿瘤表明了形态学谱。
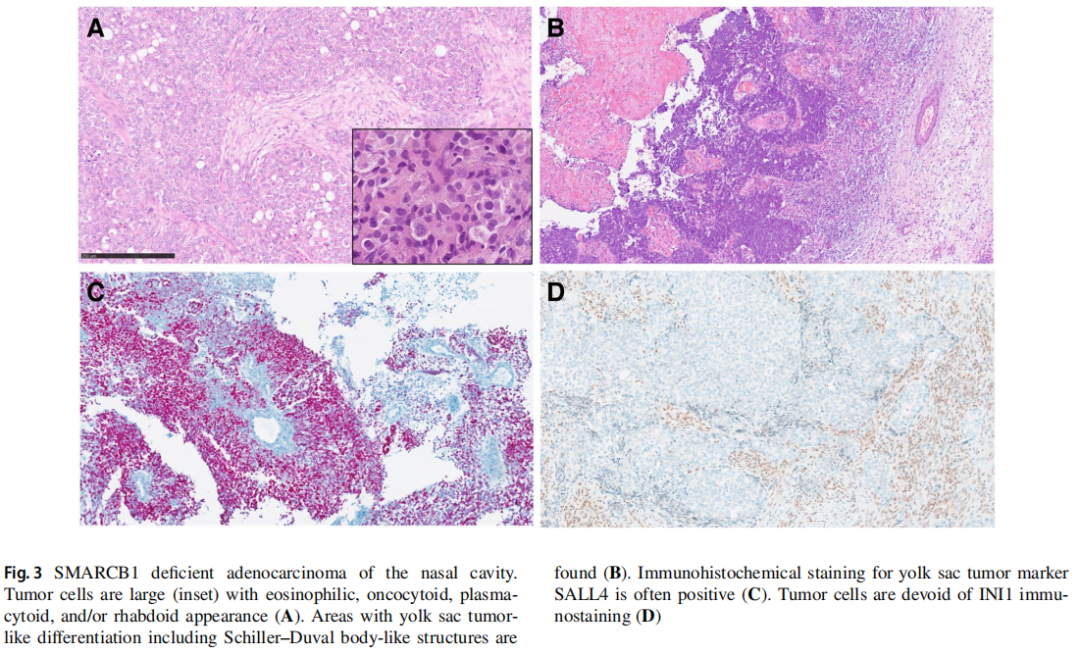
图3
SMARCA4缺陷型鼻窦癌:
自Agaimy和Weichert首次详细描述以来,文献中已经报道了不超过22例SMARCA4缺陷型鼻窦癌。这些高侵袭性肿瘤占所有未分化和低分化鼻窦癌的4%,占先前分类为SNUC 的肿瘤的9%。这个数字与档案诊断有关,而不是当前的分类系统。大多数SMARCA4缺陷型癌发生于鼻窦,并且在一部分病例中累及鼻窦的多个部位。由于其大而单调的细胞形态和常见的神经内分泌特征,最初常被误诊为神经内分泌癌。在组织学上,SMARCA4缺陷型癌为未分化癌,因此与SNUC非常相似。在稀疏到明显的反应性水肿或促结缔组织增生性间质中,它们排列成片状大的间变性上皮样细胞,形成不规则的沟通巢和小叶或小梁。
SMARCA4缺陷型畸胎癌肉瘤:
在现行的世界卫生组织分类中,这种罕见的多表型(三系)、高度侵袭性的特定部位鼻窦恶性肿瘤仍以形态学来定义,但由于其与SWI/SNF缺陷型鼻窦恶性肿瘤的分子特征存在重叠,因此在此值得提及。畸胎癌肉瘤(TCS)是指畸胎瘤样肿瘤(不同类型的胚胎上皮、神经外胚层分化和原始神经上皮)、癌样肿瘤(恶性分化的上皮成分或角蛋白阳性的低分化增殖)和肉瘤样间质/间质成分伴(主要是横纹肌母细胞和罕见的骨软骨母细胞)或不伴异源间质成分的三系生长。最近有报道称,超过80%的TCS病例中SMARCA4表达频繁缺失。然而,与上述SWI/SNF缺陷型鼻窦癌类型(SWI/SNF缺陷是定义性的)相比,鼻窦TCS仍然是基于设定的组织学标准在形态学上定义。
SWI/SNF复合体缺陷型鼻窦肿瘤通常是低分化或未分化的恶性肿瘤,具有高度侵袭性和不良预后。SMARCA4缺陷型鼻窦癌的死亡率高于该家族的其他肿瘤。
鉴别诊断具有挑战性,主要根据单个病例的组织学模式进行定义。必须考虑非角化性鳞状细胞癌(散发或HPV相关)和许多其他实体的基底细胞样变异,如NUT癌、成釉细胞瘤样尤因肉瘤和神经内分泌癌。大细胞SWI/SNF复合体缺陷型肿瘤需与SNUC、NUT罕见变异型癌、黑色素瘤、去分化脊索瘤、侵袭性间变性淋巴瘤和转移瘤等鉴别。SWI/SNF蛋白的免疫组化抗体是在适当的临床病理和形态学背景下识别这些肿瘤的有效工具。SWI/SNF复合物缺陷型恶性肿瘤可以通过免疫组织化学染色来确定,INI1抗体用于SMARCB1缺陷的恶性肿瘤,BRG1抗体用于SMARCA4 缺陷的恶性肿瘤。这些抗体是敏感的诊断工具(图3D)。
在治疗方面,关于SMARCB1缺陷型鼻窦癌和腺癌之间的区别以及两者的长期随访的现有数据有限。目前,这种区别应该能够可靠地评估两者之间的任何治疗或预后差异。患者常接受根治性手术和辅助放疗或放化疗。尽管采取了这种积极的治疗,但最近的一项多中心病例系列研究显示,在中位略超过2年的随访期间,3例患者中只有1例存活且无肿瘤。另一种治疗选择是通过多药化疗和放疗对疾病进行局部控制。迄今为止,关于SMARCB1缺陷型鼻窦癌的最大单机构研究报告了在MD Anderson癌症中心接受治疗的连续19例SMARCB1(INI-1)缺陷型鼻窦癌患者的结局。中位OS和无病生存期(DFS)分别为31.8和9.9个月。鼻腔或上颌窦肿瘤患者的疾病特异性生存率(DSS)提高84%(风险比[HR],0.136;95%置信区间[CI],0.028-0.66;p=0.005),DFS提高71%(HR,0.29;95%CI,0.097-0.84;p=0.041),高于其他鼻窦部位。接受诱导化疗的患者死于疾病的可能性降低了76%(DSS HR,0.241;95%CI,0.058-1.00;p=0.047)。
最近的研究结果表明,免疫调节剂和免疫检查点抑制剂作为SWI/SNF相关恶性肿瘤患者的潜在药物具有很好的前景。EZH2抑制剂是一种组蛋白甲基转移酶,可激活组蛋白H3赖氨酸27(H3K27me3)的甲基化,导致细胞命运相关基因的表观遗传沉默。SMARCB1缺失的肿瘤细胞表现出EZH2的组成性激活和致癌激活。EZH2抑制剂可调节肿瘤免疫原性和抗肿瘤免疫反应。
鼻窦未分化癌:
鼻窦未分化癌(SNUC)是一种没有任何分化系的高级上皮性肿瘤,只有在排除其他鼻窦和非鼻窦恶性肿瘤后才能诊断。在以前的WHO分类中,许多未分化上皮肿瘤被包括在这一术语下,直到分子病理学的进展允许将其作为独立的实体进行识别。最近,在SNUC的一个亚群中发现了IDH1/2突变,包括IDH1 R132、IDH2 R140和IDH2 R172这三个主要的热点突变。单克隆或多特异性抗体检测IDH1/2突变是一种比分子基因检测更经济的替代方法,而使用现有抗体的免疫组织化学检测IDH1/2突变缺乏全谱的能力。研究者建议同时使用免疫组织化学和分子分析。IDH蛋白质类参与Krebs循环将异柠檬酸盐转化为α-酮戊二酸。IDH突变产生一种致癌代谢物D-2-羟基戊二酸(2-HG),可诱导DNA超甲基化。
SNUCs是一种侵袭性肿瘤,临床病程隐匿,确诊时已为晚期,进展迅速,预后差。标准治疗方法曾是手术、化疗和放疗的结合,但这些治疗的最佳顺序一直存在争议。最近,一项对95例未经治疗的患者进行的具有里程碑意义的研究报告,采用诱导化疗(IC)的根治性治疗策略改善了患者结局。在接受该治疗的SNUC患者中,5年DSS为59%(95%CI,53%-66%)。对IC的反应决定了是否继续同步放化疗。对IC治疗达到完全缓解(CR)或部分缓解(PR)的反应者5年DSS为81%。无反应者接受手术和术后放疗,5年DSS为54%。
然而,在同步放化疗(CRT)的IC治疗中甚至没有达到部分缓解的患者中,5年DSS为0%。在对IC甚至没有部分缓解的患者以及接受手术加放疗或CRT治疗的患者中,5年DSS为39%(校正风险比为5.68[95%CI,2.89-9.36])。作者的结论是,在对IC有良好反应的患者中,与接受确定性手术的患者相比,确定性CRT可改善生存。对于对IC没有良好反应的患者,可行时手术似乎提供了更好的疾病控制机会和提高生存率。
IDH2突变的SNUCs预后优于其他SNUCs。IDH突变的存在提供了其他治疗选择,包括选择性小分子抑制剂(如靶向IDH2突变的恩西地平和靶向IDH1突变的艾伏尼布),这些抑制剂可抑制DNA超甲基化,导致癌细胞生长延迟和诱导细胞分化。
HPV相关的多表型鼻窦癌:
HPV相关的多表型鼻窦癌(HMSC)是一种几乎完全局限于鼻窦的上皮性恶性肿瘤,且含有高危型HPV。最常见的血清型是33型,其次是其他罕见的血清型,如52型、56型等。HMSC免疫组化p16呈阳性,使用RNA原位杂交等直接检测的高危HPV检测也呈阳性。阴性的p16结果有助于排除这种肿瘤类型,但阳性的p16可能是非特异性的,不能作为这种肿瘤的HPV替代指标。HMSC在组织学上具有高度多形性,可以模拟不同的涎腺和非涎腺肿瘤类型。HMSCs的特征是导管上皮细胞和肌上皮细胞的双重群体,主要是实性生长方式,坏死区域,以及覆盖的表面上皮类似于高度异型增生。在某些地方,HMSC的组织学可能与腺样囊性癌(AdCC)相似,这是一个重要的鉴别诊断,因为与真正的AdCC不同,HMSC通常预后良好。HMSC的组织学表现通常与高级别细胞形态学、破坏性生长和局部复发倾向相关。尽管具有侵袭性的外表,HMSC具有较低的转移潜能和很少的致死性行为倾向。
鼻窦腺样囊性癌
腺样囊性癌(AdCC)是一种由上皮和肌上皮肿瘤细胞组成的侵袭性恶性肿瘤,肿瘤细胞排列成管状、筛状和实性,伴有嗜酸性细胞外基质和重叠的基底膜材料,并常伴有MYB、MYBL1和NFIB基因的融合。AdCC的基因组标志物是t(6;9)或t(8;9)易位,分别导致MYB::NFIB和MYBL1::NFIB融合(图4A和B)。前者的变异见于>80%的病例,后者见于大约5%的病例。基因融合或其他机制导致的MYB/MYBL1激活是AdCC发病机制中的关键事件。1p、6q和15q的缺失与高级别肿瘤相关,而14q的缺失仅见于低级别肿瘤。

图4
异型AdCC最近被认为是AdCC的一种形态学变异,好发于鼻窦黏膜。异型AdCC表现出不同寻常的形态,包括鳞状分化和大囊性生长(图4C)。异型AdCC的另一种不同寻常的组织学模式是显著的肾小管嗜酸性粒细胞增多和管腔细胞突出,这与绝大多数以肌上皮细胞为主的基底样AdCC形成了对比(图4D)。
AdCC的二代测序在FGF/IGF/PI3K、染色质重塑和NOTCH信号通路的基因中发现了大多数发生率较低的突变。对于晚期、复发或转移性AdCC患者,传统的治疗药物在延长生存期方面效果不佳。因此,为了开发可改善结局的靶向疗法,人们付出了大量努力。特别是,在AdCC中存在NOTCH变异被证明与不良的生存率相关。有这些变异的患者可能从抗 NOTCH药物(如Brontictuzumab)中获益。在同一细胞系中,AL101(如Brontictuzumab)已被证明在具有激活NOTCH1突变的AdCC的体外和体内模型(AdCC细胞系、类器官和患者来源的异种移植模型)中具有强效抗肿瘤作用。
在最近的一项研究中,B7家族成员B7-H4(VTCN1)的高表达与AdCC较差的预后相关,无论临床分期和组织学亚型如何。B7-H4在实性ACC中高表达,是该疾病的独立预后标志物。MD安德森癌症中心最近的另一项研究表明,在各种AdCC亚型中,多种可采取措施的蛋白/通路改变的共同发生表明,对于这种研究不足的异质性疾病,独特的治疗弱点和最佳联合治疗的机会。
好发于鼻窦的软组织肿瘤
具有鉴别诊断意义的分子变异:
头颈部可能是各种软组织肿瘤的宿主,但对所有这些实体的讨论超出了这篇综述的范围。因此,本文只纳入了鼻窦经常发生或特有的软组织恶性肿瘤,并有诊断有用的分子变异。所讨论的实体列在表2中。


表2
双表型鼻窦肉瘤:
双表型鼻窦肉瘤(BSS)是一种低级别恶性的梭形细胞间叶性肿瘤,具有神经源性和肌源性分化,仅见于鼻窦区域。BSS的分子定义是PAX3基因的重排,它参与神经源性、黑素细胞性和骨骼肌分化(图5A-D)。在超过一半的病例中,最常见的基因融合是PAX3::MAML3,其他融合伴侣包括NCOA1、NCOA2、FOXO1、FOXO6和WWTR1。
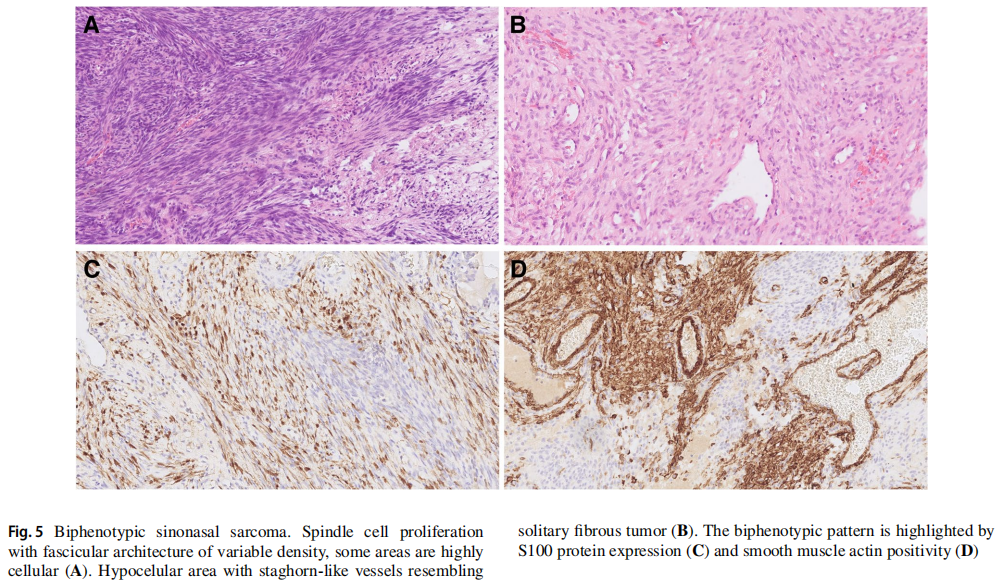
图5
BSS通常是一种低级别肿瘤,据报道有1/3到一半的病例有局部复发,并且可能在诊断后很多年复发。目前已发表3例BSS高级别转化病例,其中1例发展为横纹肌肉瘤(RMS)。BSS和RMS的分子检测显示相似的基因融合,包括PAX3::FOXO1和PAX3::NCOA1。BSS和RMS可能形成一个病变的连续体。
外间充质软骨粘液样肿瘤:
外间充质软骨粘液样肿瘤(ECT)(又称RREB1::MRTFB重排肿瘤)是一种恶性潜能不确定的肿瘤,主要发生于舌,极少发生于舌外部位。免疫表型无特异性,分子基因学检测是诊断ECT的首选方法。该肿瘤在大多数病例中以RREB1::MRTFB融合为特征,而在少部分病例中存在EWSR1基因重排。ECTs在基因学和组织学上与软组织肌上皮肿瘤相关,可能被误认为其他梭形细胞形态占优势的间叶性肿瘤。ECT通常是良性的,没有转移。手术在大多数情况下是治愈的,尽管可能发生局部复发。最近,携带RREB1::MRTFB融合基因的肿瘤在不同的头颈部和非头颈部都有报道,这些肿瘤表现出不同的形态学和免疫表型,并与舌体ECT重叠。值得注意的是,在文献中报道的10例病例中,有5例累及咽旁间隙和下颌骨/或鼻窦道。据报道,携带RREB1::MRTFB融合的鼻窦肿瘤更类似于鼻窦软骨间充质错构瘤,但缺乏大多数鼻窦软骨间充质错构瘤所具有的DICER1变异。
GLI1变异的软组织肿瘤:
GLI1变异的软组织肿瘤最近被描述为组织来源不明的间叶性肿瘤,其特征为上皮样或球样(以及较少发生的局灶性梭形)形态和非特异性免疫特征,发生于头颈部的病例占40%。在2/3的病例中,这些肿瘤存在GLI1融合,包括ACTB::GLI1、PTCH1::GLI1、MALAT1::GLI1和DERA::GLI1,而其余的肿瘤存在GLI1扩增。GLI1的各种融合伴侣最好通过靶向RNA测序确定,GLI1扩增也可以通过FISH检测。必须考虑到12号染色体上相邻基因(尤其是CDK4、MDM2、STAT6和DDIT3)共扩增并被FISH检测到的可能性。此外,GLI1免疫染色对GLI1重排的间叶肿瘤具有较高的特异性和良好的敏感性。GLI1 IHC和p16染色相结合在检测GLI1扩增肿瘤方面具有优势。
GLI1变异的软组织肿瘤的生物学行为从完全惰性到潜在的侵袭性转移肿瘤不等。大约20%的病例会出现局部复发或远处扩散。GLI1变异的肿瘤的潜在靶向治疗方案包括sonic hedgehog信号通路抑制剂。
鼻窦横纹肌肉瘤:
横纹肌肉瘤(RMS)是一种具有临床、预后和生物学异质性的肿瘤,在形态学上可分为四种主要亚型。然而,分子基因学研究结果描述了6种不同的亚型:具有未知驱动突变/融合的胚胎型RMS、具有FOXO1融合的肺泡型RMS、具有MYOD1激活突变的MYOD1突变型RMS、VGLL2/VGLL3/NCOA2重排的RMS、具有复杂基因背景的多形性RMS,以及具有EWSR1或FUS融合伴侣的TFCP2重排RMS。梭形细胞/硬化型RMS通常细胞角蛋白和成肌标志物阳性(至少局灶性),主要是MyoD1、myogenin、desmin或PAX7。pankeratin、ALK和desmin联合阳性高度提示TFCP2融合RMS。
VGLL2/VGLL3/NCOA2基因重排的RMS属于梭形细胞/硬化型RMS,好发于新生儿或婴儿,并且好发于头颈部。特征分子基因事件涉及VGLL2::CITED3、VGLL2::NCOA2、TEAD1::NCOA2或SRF::NCOA2基因融合。最近报道了6例以TCF12、EP300和PPARGC1A作为融合伴侣的新型VGLL3重排病例。临床上,这些肿瘤预后良好。仅报告了4例转移病例。复发时,其中3例表现出高级别形态,并伴有细胞周期蛋白编码基因(特别是TP53、CDKN2A/B和FGFR4)突变。完全手术切除通常是治愈性的,而对于不可切除的病例,由于有可能发生高级别转化,建议采用RMS类型的化疗。
TFCP2重排RMS(TFCP2-RMS)是一种具有横纹肌分化的梭形细胞/硬化性和非常侵袭性的间叶性肿瘤,以EWSR1/FUS::TFCP2重排为特征。在一部分病例中,已经描述了ALK基因的半合子缺失或扩增。TFCP2-RMS肿瘤好发于年轻人的颌骨和颅骨,常继发软组织受累。这种肿瘤通常在诊断时已处于晚期,病程迅速,尽管进行了积极的多模式治疗,但预后极差,3年总生存率为28%。治疗方案包括手术,如果在诊断时已发生大规模肿瘤扩散,则可能不适合手术。化疗和放疗不能延长生存期。放疗联合ALK抑制剂(克唑替尼、阿来替尼、洛拉替尼和/或帕唑帕尼)在个别病例中显示出部分缓解。
成釉细胞瘤样尤因肉瘤:
成釉细胞瘤样尤因肉瘤(ALES)是尤因肉瘤(ES)的一种有争议的变体,其定义为存在t(11;22)和EWSR1::FLI1融合。由于上皮标志物(泛细胞角蛋白和p63/p40)和ES相关标志物(CD99和NKX2.2)通常表达,因此推测它是否代表上皮性肿瘤或间质性肿瘤。目前尚不确定该肿瘤应按照癌还是肉瘤治疗方案进行治疗。根据ES特定方案,许多肿瘤可以通过手术和辅助化疗来治疗。
EWSR1/FUS::POU2AF3(COLCA2)肉瘤:
由Agaimy等人首先报道的EWSR1/FUS::POU2AF3(COLCA2)肉瘤是一种新认识的侵袭性肿瘤,尽管采用多模式治疗,但仍表现出局部复发和转移扩散的倾向。该肿瘤累及成人患者,通常发生于头颈部,尤其好发于鼻窦。其形态谱从具有轻度核异型性的梭形细胞增殖,到由具有神经内分泌分化特征的梭形细胞和圆形细胞组成的片状和束状双相肿瘤,再到具有高核级别的纯圆形细胞肿瘤(图6A-D)。此外,一些肿瘤被报道含有腺体、横纹肌细胞或成骨分化灶。大约一半的病例显示泛细胞角蛋白阳性,可能导致与滑膜肉瘤的诊断混淆。一般而言,EWSR1与不同伴侣的融合在软组织肿瘤中很常见,包括滑膜肉瘤中替代性EWSR1::SSX1融合的罕见实体,或者潜在的FUS与POU2AF3融合,而非EWSR1。因此,FISH检测EWSR1断裂并不是充分的诊断性检测。相反,建议通过靶向RNA测序做出正确诊断。
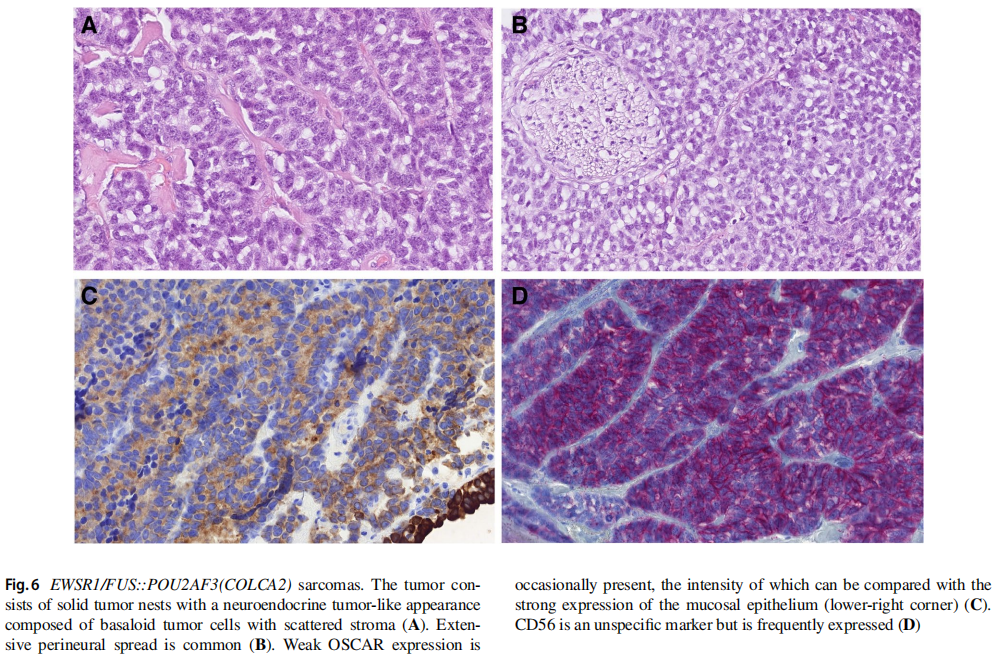
图6
综上所述,随着分子检测的广泛应用,头颈部肿瘤的分类在总体上有了快速的发展,在鼻窦区和前颅底肿瘤领域也明显得到了发展。越来越多的免疫组化替代物的出现为病理学家提供了快速识别诊断性分子异常的工具,而对于临床医师而言,这些工具越来越多地帮助他们选择适当的靶向治疗。虽然诊断仍主要依赖组织学和免疫组织化学,但对鼻窦肿瘤分子基础的新认识显然使病理学家能够提高诊断的准确性,从而更好地进行微调和消除重叠诊断。可以说,WHO第5版分类采用了鼻窦恶性肿瘤的分子生物学基础,反映了这些技术在头颈肿瘤科室日常实践中所取得的不可逆转的地位。
参考文献:
Skálová, Alena et al. “Molecularly defined sinonasal malignancies: an overview with focus on the current WHO classification and recently described provisional entities.” Virchows Archiv : an international journal of pathology, 10.1007/s00428-024-03775-y. 16 Mar. 2024, doi:10.1007/s00428-024-03775-y

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#GLI1# #鼻窦恶性肿瘤# #鼻窦道#
6