
Nat Genet:基于三个大型生物库的测序数据进行泛祖先人类疾病罕见编码变异分析
2024-09-26 测序中国 测序中国 发表于上海

研究团队利用三个大型生物库测序数据创建了人类疾病罕见编码变异关联数据集,并评估了不同祖先组成在罕见变异关联分析中的作用。
近年来,大规模测序的应用促进了人们对罕见编码变异在人类表型变异性中作用的了解。然而,此前研究在统计方法或者检测能力层面仍存在局限性,例如不考虑群体祖先和其他协变量,或者对高度不平衡表型产生错误校准的测试统计的模型等。
目前,一些生物库倡议已优先纳入来自未充分研究群体的样本,目标是增加对健康和疾病的无偏颇认识。其中,All of Us研究项目(AoU)已经完成了美国近25万名参与者的全基因组测序(WGS),其中约一半是非欧洲遗传祖先。
近日,美国麻省理工学院和哈佛大学Broad研究所的研究团队在Nature Genetics发表了题为“Rare coding variant analysis for human diseases across biobanks and ancestries”的文章。研究团队利用三个大型生物库测序数据创建了人类疾病罕见编码变异关联数据集,并评估了不同祖先组成在罕见变异关联分析中的作用。总之,该泛祖先分析对人类疾病关联测序中的多祖先和跨生物库研究具有重要启示意义。

文章发表在Nature Genetics
主要研究内容
研究人员结合来自UK Biobank(UKB)和Mass General Brigham Biobank (MGB)的大规模全外显子组序列(WES)数据,以及来自AoU的WGS数据进行了泛祖先分析。经过质量控制程序,共有748879人纳入后续深入分析。根据与千人基因组计划样本的遗传相似性将个体分为不同群体,即非洲(AFR)、东亚(EAS)、欧洲(EUR)、南亚(SAS)和混血(AMR)祖源。与预期一致,AoU的祖先多样性最高,49.9%的参与者具有除EUR以外的遗传决定的祖先;相比之下,来自UKB和MGB的94.4%和83.5%的样本在遗传上被确定为欧洲血统。
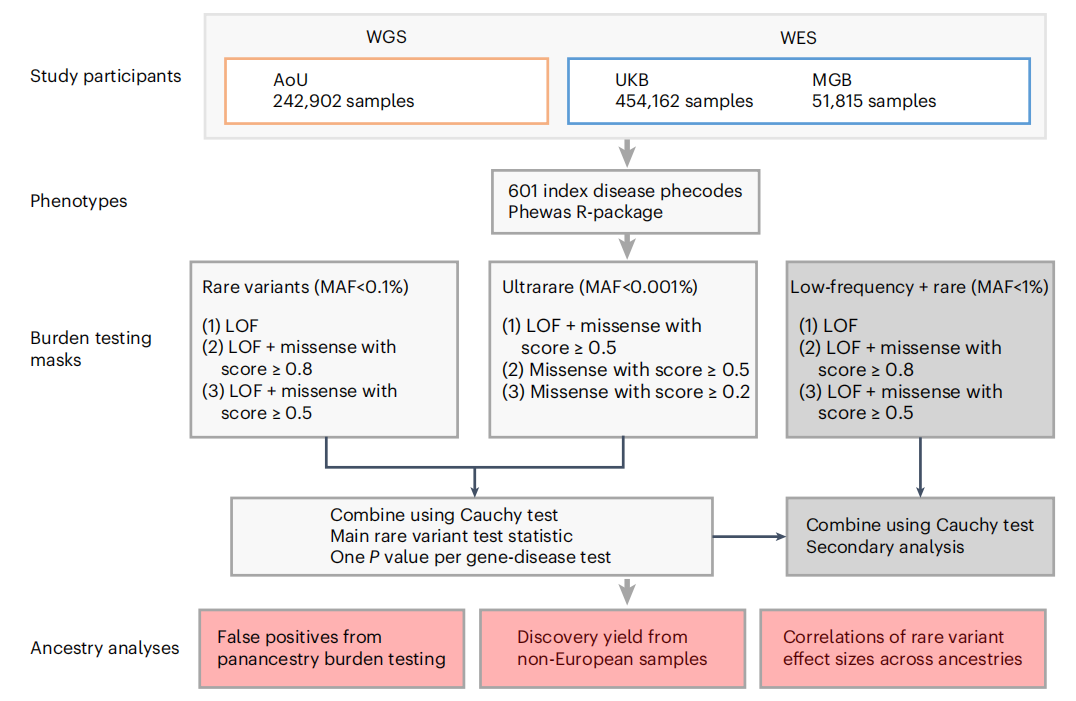
图1. 人类罕见变异分析整体流程
跨生物库的罕见变异分析
为了对主要遗传分析定义疾病端点,研究人员从国际疾病分类代码映射中创建了多达1866个编码(疾病表型),并使用分层聚类算法将其精简为601个索引编码。对于601个编码端点,研究评估了6种罕见变异模块,包括功能缺失(LOF)变异和错配变异的各种组合。
当评估单个数据集时,在UKB中观察到的显著关联数量最多,MGB的关联最小。在多祖先Meta分析中,11060516对独特的基因-表型对中的363个关联在总体FDR小于0.01时达到显著性,包括165个独特表型和123个独特基因。
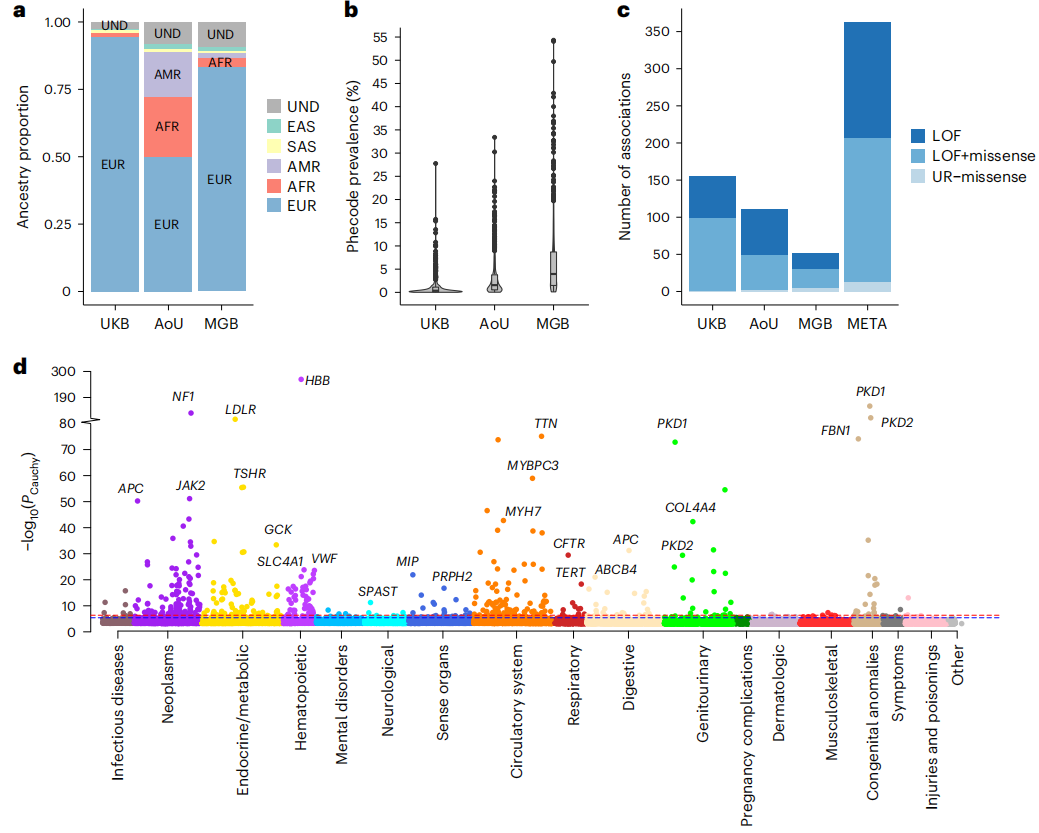
图2. 罕见变异的多祖先Meta分析
核心基因对人类疾病表型的遗传效应
在Meta分析的363个显著关联中,301个直接在人类OMIM数据库中被报道,或者与该数据库中的条目有可能相关。这些分析结果突出了多效性疾病基因(即与多种疾病结局和后遗症相关的基因)和大效应量相关的基因,均指向人类疾病表型的核心基因。其中包括FBN1-马凡综合征的致病基因,其与心血管和遗传疾病代码中的13种疾病的关联,FBN1对“染色体异常”和“遗传疾病”的影响最大;在常染色体多囊肾病的致病基因PKD1-a中发现了最多的基于基因的关联,PKD1与29个编码相关,最明显的是泌尿生殖系统先天性异常和慢性肾功能衰竭。
此外,研究还鉴定了许多孟德尔基因-疾病联系,这在此前的研究中并没有被观察到。例如,PTEN与几种考登综合征的表型相关,包括先天性异常和甲状腺疾病;LMNA和TNNT2与心肌病和各种后遗症有关;CFTR与囊性纤维化相关;SMAD3、COL3A1和LDLR与血管动脉瘤相关。总之,以上结果突出了测序数据的持续增长能够增加对疾病更深刻地认识。
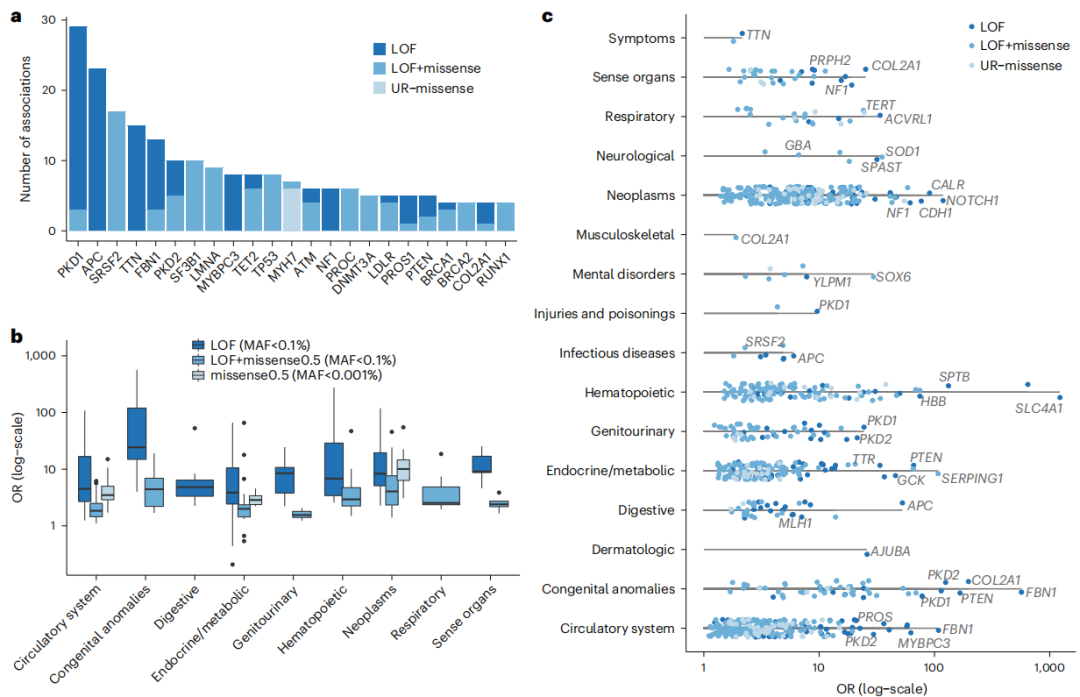
图3. 遗传效应大小和多效性关联确定人类疾病表型的核心基因
跨祖先罕见变异效应的一致性
最后,研究团队探讨了罕见编码变异对人类疾病的影响是否在祖先之间是一致的,以及欧洲血统样本和其他遗传祖先个体之间的影响是否一致。首先,研究人员通过对来自UKB和MGB的EUR个体的Meta分析确定了具有启发性的显著信号,然后进一步评估这些信号在不同AoU数据集中的效应大小。
结果发现,来自欧洲祖先样本的显著效应大小与来自其他祖先的估计效应具有良好的相关性,类似于关于共同变异和数量性状的跨祖先研究结果。此外,研究团队还进行了一些敏感性分析,包括去除与年龄或白血病结果相关的基因,并对有效样本量进行分析,这些分析结果基本一致。
总的来说,该研究结果证明罕见LOF变异的效应大小在EUR和其他遗传祖先之间具有合理的一致性,表明进一步的跨祖先分析方法可以提高疾病测序关联研究的鉴定能力,并且因果变异在不同祖先之间的影响具有高度一致性。
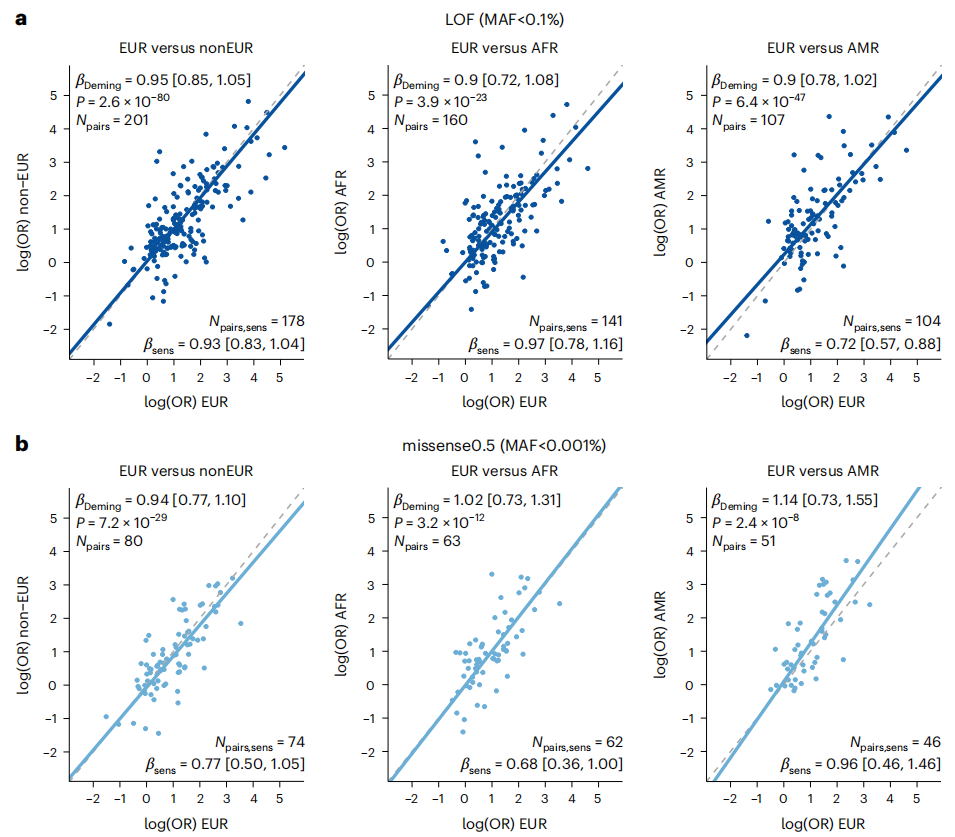
图4. 罕见疾病编码变异效应大小在EUR和其他遗传祖先之间的相关性分析
结 语
综上所述,该研究对来自三个大型生物库的测序数据进行了泛祖先分析,包括AoU项目的数据。基于混合效应模型,研究人员对748879名个体的601种疾病进行了罕见变异检测,其中包括155236名与欧洲血统不同的个体。最终,研究团队确定了363个显著关联,这些关联突出了人类疾病表型的核心基因,并确定了潜在的新关联。该研究提出了一个罕见变异关联数据集,涵盖了广泛的人类疾病表型,为跨祖先人类疾病的罕见编码变异的一致影响提供了新见解。
研究团队还发布了一个门户网站(https://hugeamp.org:8000/research.html?pageid=600_traits_app_home),用户可以浏览来自单个数据集(UKB,AoU和MGB)、各种Meta分析、不同祖先的比较研究结果等。
论文原文:
Jurgens, S.J., Wang, X., Choi, S.H. et al. Rare coding variant analysis for human diseases across biobanks and ancestries. Nat Genet (2024).
https://www.nature.com/articles/s41588-024-01894-5
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言










#测序# #生物库# #罕见编码#
86