
Nat Commun:山东大学董婷等团队合作研究揭示了MFSD7C通过抑制铁死亡来保护溶血诱导的肺损伤
2024-09-23 iNature iNature 发表于上海

该研究发现提供了溶血性并发症和铁死亡之间的详细联系,为溶血性疾病患者提供了潜在的治疗靶点。
溶血导致对肺损伤的易感性,并预测疟疾和镰状细胞病(SCD)等疾病的不良预后。然而,潜在的病理机制尚不清楚。
2024年9月19日,山东大学董婷、娄红祥,中国医学科学院刘晓辉共同通讯在Nature Communications 在线发表题为“MFSD7C protects hemolysis-induced lung impairments by inhibiting ferroptosis”的研究论文,该研究发现提供了溶血性并发症和铁死亡之间的详细联系,为溶血性疾病患者提供了潜在的治疗靶点。
研究阐述了含有7C的主要促进剂超家族结构域(MFSD7C)通过防止铁死亡来保护肺免受溶血性损伤。机制上,HuLEC-5A细胞MFSD7C缺乏导致线粒体功能障碍、脂质重塑、ACSL4和GPX4失调,从而增强脂质过氧化,促进铁死亡。此外,系统递送药MFSD7C mRNA负载纳米颗粒可有效预防溶血性小鼠(如HbSS-Townes小鼠和PHZ攻击的7C-/-小鼠)的肺损伤。

溶血是指血液循环中红细胞的破坏,是各种疾病的主要病理并发症之一,包括镰状细胞病(SCD)、疟疾、自身免疫性疾病和骨髓衰竭。在稳态条件下,衰老红细胞的破坏导致血管内环境中游离血红蛋白、血红素和铁的释放。过量的血红素被血红素中和及内化,并在细胞内通过血红素加氧酶(HMOX1)降解产生胆绿素、一氧化碳和亚铁。溶血疾病中广泛的溶血破坏了血红素和铁的体内平衡,使它们的清道夫分子饱和。血红素和铁调节失败导致氧化应激和炎症,导致不可逆转的细胞和组织损伤或细胞死亡。具体来说,在溶血过程中,不受控制的铁释放到循环中会引发正常和无核细胞(如血小板)的铁死亡。
铁死亡是一种铁依赖性的细胞死亡形式,由广泛的铁积累和脂质过氧化引起。虽然多不饱和脂肪酸(PUFAs)的插入增强了膜的流动性,但PUFAs的过量氧化可能导致细胞氧化危机。由于肺部疾病患者的铁或氧化还原稳态异常,铁死亡被认为参与肺部疾病。例如,急性肺损伤小鼠表现出铁超载、谷胱甘肽(GSH)消耗和丙二醛(MDA)积累、肺组织SLC7A11和GPX4表达水平下调等铁致凋亡生理特征。同样,在慢性破坏性肺部疾病中,暴露于香烟烟雾的支气管上皮细胞表现出内皮网应激和线粒体功能障碍,并伴有GPX4和GSH水平不足,导致铁积累和铁死亡。此外,铁死亡在肺纤维化中也有病理作用,如脂质过氧化和ROS产生增加,导致成纤维细胞分化和激活,以响应Erastin。然而,铁死亡诱导肺损伤的详细机制需要更多的重点研究。
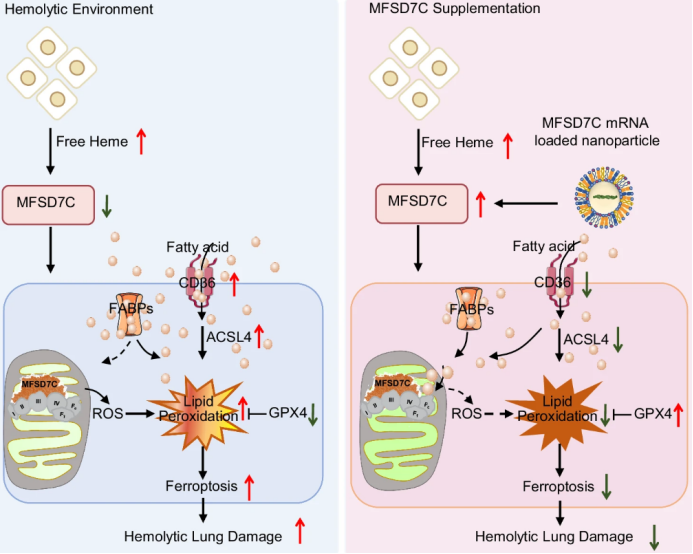
MFSD7C通过抑制铁死亡来保护溶血诱导的肺损伤(图源自Nature Communications )
含有7C的主要促进物超家族结构域(MFSD7C),也被称为FLVCR2和SLC49A2,最初被确定在巨噬细胞中介导血红素铁转运。然而,进一步的研究没有观察到MFSD7C过表达和敲除小鼠血红素水平的变化。最近一项研究报道,MFSD7C是血脑屏障上的胆碱转运蛋白,在细胞中表达MFSD7C可显著增加胆碱摄取。之前的研究表明,MFSD7C在巨噬细胞中响应血红素水平,通过将ATP合成转换为产热来调节线粒体能量代谢。然而,MFSD7C在铁死亡和溶血相关肺损伤中的作用从未被研究过。
目前研究了MFSD7C在溶血小鼠模型肺损伤中的作用及其潜在机制。研究发现MFSD7C通过稳定线粒体功能和调节GPX4和ACSL4的表达来防止脂质过氧化,从而保护肺细胞免于铁死亡。MFSD7C基因敲除通过促进肺铁死亡加重肺部炎症和肺纤维化。载体MFSD7C mRNA纳米颗粒可有效改善溶血小鼠肺损伤模型。
参考消息:
https://www.nature.com/articles/s41467-024-52537-6
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言











#肺损伤# #溶血# #MFSD7C#
47