硝酸甘油用于治疗心肌梗死患者——风险与益处综述
2012-01-01 胡世红 广西柳州市人民医院

急性心肌梗死(AMI)及其后果是全球发病率和死亡率的首位原因。硝酸甘油(GTN)仍然是心绞痛和AMI的一线治疗。GTN的益处是通过产生氧化氮(NO)引起血管扩张并增加心肌血流来实现的。然而,持续给予GTN则引起耐药,限制了该药的应用。GTN耐药至少部分是由乙醛脱氢酶2(ALDH2,一种可将GTN转化为血管扩张剂NO的酶)的失活引起的。最近我们发现在动物MI模型
急性心肌梗死(AMI)及其后果是全球发病率和死亡率的首位原因。硝酸甘油(GTN)仍然是心绞痛和AMI的一线治疗。GTN的益处是通过产生氧化氮(NO)引起血管扩张并增加心肌血流来实现的。然而,持续给予GTN则引起耐药,限制了该药的应用。GTN耐药至少部分是由乙醛脱氢酶2(ALDH2,一种可将GTN转化为血管扩张剂NO的酶)的失活引起的。最近我们发现在动物MI模型中,除了GTN对血管床的作用外,持续的治疗可负面影响缺血后的心肌活力,从而引起梗死面积增大。醛缩酶-1(ALDA-1,一种ALDH2激活剂)与GTN联用,可改善反应性醛加合物的代谢并可预防MI后GTN引起的心脏功能不全。在本综述中,我们将描述GTN用于MI的益处和风险相关的分子机制。
硝酸甘油的有益作用
尽管药物治疗有了很大进展,但缺血性心脏病和AMI仍然是全球发病率和死亡率的主要原因。根据WHO的资料,每年有7百多万人死于缺血性心脏病。因此,需要开发新的药物和非药物对策以造福MI患者。GTN自150年前发现以来,一直是不稳定性心绞痛、MI和心衰患者最常用的治疗。GTN促进血管扩张和耐受的能力,早在20世纪GTN工业上升时代就已受到明确注意。工厂的工人,通常暴露于高水平的有机硝酸酯,经常主诉星期一头痛,而在周末头痛消失。其实,患有心绞痛或心衰的工人,经常感觉到工作周胸痛缓解,但在周末又复发。原自GTN血管扩张作用的这两种影响,迅速引起了医生的注意。硝酸酯耐受现象,因“星期一病”的发作和硝酸酯撤药/过度代偿的“星期天心脏发作”而得到极好地认识。
GTN的正面作用来自其促进血管扩张的能力,引起心脏血流量的增加。GTN对全身静脉的扩张作用也是明显的,可降低前负荷和进一步降低心室壁张力。GTN对恢复缺血心脏的氧气和营养供-需平衡是极为有效的。然而,持续的GTN应用可引起耐药,并与促氧化作用、内皮功能不全和对缩血管剂的敏感性增高有关。在临床实践中应用的其它硝酸酯包括硝酸异山梨酯,其活性代谢物单硝酸异山梨酯、季戊四醇四硝酸酯、erythrytyl-tetranitrate和尼可地尔。
GTN治疗性应用的历史
1847年,在都灵Theophile-Jules Pelouze’s实验室工作的Ascanio Sobrero发现了GTN并首先注意到由GTN所致的剧烈头痛可持续数小时。两年后,德国科学家Constantin Hering得知了Sobrero的头痛报告,便在健康志愿者中试验GTN,并精确地观察到可引起头痛。1851年Alfred Nobel参与了合成GTN并认识到GTN的科学和金融潜能。此后几年,他开始在瑞典制造GTN。Nobel在其一生中长时间健康很差,患有剧烈疼痛和心绞痛。因此,具有讽刺意味的是,1890年他的医生建议用GTN治疗他的心脏病。19世纪的后50年,几位英国科学家对新发现的被认为是强效血管扩张剂二戊基硝酸酯感兴趣。Lauder Brunton在1867年应用这种化合物来缓解心绞痛,并首次报道多次剂量后出现耐药。接着Brunton的工作,科学家们集中于记录硝酸酯类化合物对几种病理体系包括心绞痛、MI、高血压和心力衰竭的效果。最后,在19世纪末GTN被确立为缓解心绞痛的治疗。然而,GTN引起益处的机制只是在80年后才被发现。
在20世纪70年代后期,发现GTN的血管扩张作用是由氧化氮介导的,而NO显然是从血管平滑肌内的GTN产生的。几年后,发现哺乳动物细胞可合成NO。1998年,在Alfred Nobel发明炸药并首次观察到GTN临床效果后约130年,医学或生理学Nobel奖因“氧化氮是心血管系统的信号分子”被授予了Robert Furchgott, Louis Ignarro 和Ferid Murad. GTN仍然是缓解心绞痛的首选治疗,其它有机硝酸酯和无机硝酸酯也被应用,但GTN迅速起效及其已确立的效果,使它成为缓解心绞痛的支柱。
在硝酸甘油生物活性中的ALDH2
GTN的血管扩张作用作为由NO介导的一个过程被发现。随后的研究发现,GTN(或其它硝基化合物)和硫醇之间的化学反应,产生一种中间体S-亚硝基硫醇,可引起NO进一步产生。如今,常常推测GTN是在平滑肌细胞中转化成NO,激活可溶性鸟苷酸环化酶产生cGMP,后者又引起血管平滑肌松弛(图1)。尽管进行了强化的基础和临床研究,但从GTN产生NO的分子机制仍有待阐明。事实上,已经提出在GTN的生物活性中可能涉及到1种或多种酶促反应。由于GTN的生物活性是由非酶反应或酶促反应之一所介导的,故这种研究是很复杂的。内源性还原剂(即硫醇和抗坏血酸)可引起非酶GTN活性,然而,其对GTN介导的血管扩张的贡献似乎是有限的。对GTN的生物活性也涉及到几种酶促反应,包括谷胱甘肽-S-转移酶、黄嘌呤氧化还原酶和细胞色素P450系统。
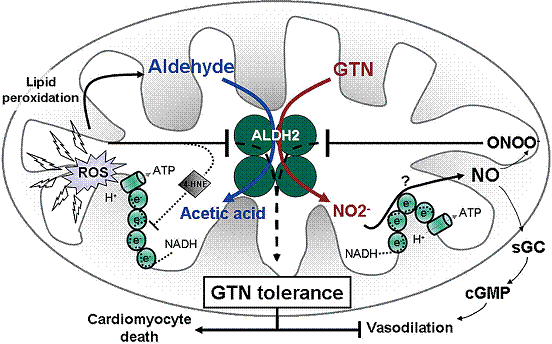
对GTN的耐药主要是由ALDH2还原酶活性的氧化失活引起的,可导致GTN生物活性降低、亚硝酸盐和NO水平降低。因为可溶性鸟苷酸环化酶活性和血管平滑肌cGMP产生不足,这种反应可消除GTN引起的血管扩张现象。除了其还原酶活性外,ALDH2的脱氢酶活性也因为GTN耐受而下调,导致线粒体内毒性乙醛的积累,进一步破坏线粒体的呼吸链,并引起活性氧产生。GTN介导的ALDH2失活激发心肌细胞凋亡,以及消除GTN对血管平滑肌的扩张作用。
然而,因为这些酶没有一个能催化从GTN选择性地形成1,2-甘油基二亚硝酸盐(1,2-GDN),故人们继续寻找GTN的代谢酶。2004年,Stamler等鉴定出线粒体ALDH2是催化GTN生物转化为1,2-GDN的关键酶。ALDH2是一种线粒体酶并且是随不同的组织分布表达的、由17种同功酶组成的、NAD(P)+依赖的ALDH家族的成员之一。ALDH2是一种四聚体酶,已知其主要作用是乙醇代谢,催化乙醛氧化形成乙酸。然而,除了其脱氢酶活性外,ALDH2至少在体外还有酯酶和还原酶活性。基于ALDH2的酯酶活性,与GTN反应预期能产生一种能被催化产生硝酸盐的S-亚硝基硫醇中间产物。然而,亚硝酸盐的形成表明S-亚硝酸基硫醇已减少并不被水解。因此,提示硫醇通过活性S-亚硝酸硫醇的形成增强GTN的活性。事实上,纯化的ALDH2只在较低的浓度才催化1,2- GDN形成。作为GTN还原酶以及促进GTN诱导血管扩张的ALDH2,其作用在过去10年已得到广泛证明。纯化的线粒体ALDH2主要从低水平的GTN(1 μmol/L)催化1,2-GDN形成。然而,在烟酰胺腺嘌呤二核苷酸(NAD+)存在时,GTN的代谢率以及1,2-GDN/1,3-GDN比值,在从牛肝提纯的ALDH2或过表达的人ALDH2中显著增加。使用不同的动物模型,已经显示用水合氯醛、戒酒硫或氰氨抑制ALDH2,可减弱静脉内注射GTN引起的低血压。用ALDH2或底物处理主动脉环可抑制GTN引起的血管扩张。有趣的是,这些抑制剂不能减弱硝普钠引起的血管松弛(直接激活鸟苷酸环化酶),提示GTN介导的作用主要是对血管ALDH2活性的特殊抑制。在敲出了ALDH2的老鼠,小剂量GTN输注的降压作用被消除,而大剂量输注时降压作用显著降低。正如所预期的,GTN对1,2-GDN的生物活性主要是在敲出了ALDH2的老鼠被消除。
总之,所有这些研究均支持ALDH2对GTN生物活性的重要作用。ALDH2在催化部位含有3个半胱胺酸残基(Cys 301, 302 和 303)。Stamler等根据Cys 302(参与催化的半胱胺酸)与2个邻近的半胱胺酸残基之一之间的二硫化物形成,对ALDH2还原酶活性提出了一种特别的机制。因此,Mayer等提出GTN脱硝基和生物活性反应了ALDH2催化GTN生物转化的两条分别的通路,两条通路都涉及到作为起始反应步骤在Cys302形成thionitrate中间产物。然而,以这部分为基础的引起ALDH2催化GTN生物转化的分子机制仍然不明。
由线粒体ALDH2的GTN还原酶活性产生的亚硝酸盐进一步被还原产生NO,NO激活可溶性鸟苷酸环化酶并促进血管扩张,这一假说已被广泛接受(图1)。然而,亚硝酸盐转化成NO的分子机制仍然有争论。Chen等证明,用取自成纤维细胞的孤立线粒体与不同浓度的GTN温育可引起剂量依赖NO产生,而ALDH2抑制可阻止这种反应。已有报道哺乳动物的线粒体存在一种与线粒体呼吸链III 和 IV复合体相关的亚硝酸还原酶活性。然而,对GTN3-电子还原产生NO的机制仍然不明。最近的研究证实在GTN衍生亚硝酸盐的生物反应中并未涉及到线粒体呼吸链。

醛缩酶-1 (一种选择性ALDH2活化剂)增强ALDH2的还原酶活性。醛缩酶-1对ALDH2还原酶活性的影响目前尚未明确。
GTN耐药涉及的机制
GTN对血管平滑肌的生物活性,对促进心绞痛和充血性心衰有效的血管松弛治疗是必需的。然而,GTN的用途广泛受到耐药发生的限制。持久的GTN耐药至少部分可致GTN生物活性受损。然而,GTN治疗可引起交叉耐药,可致其它硝基血管扩张剂的松弛作用受损。尽管其因素很多,包括可溶性鸟苷酸环化酶脱敏、cGMP-依赖的激酶和肌球蛋白轻链磷酸化,GTN耐药主要是由于线粒体内的ALDH2不能催化GTN转化为1,2-GDN和亚硝酸盐所介导的。例如,水合氯醛,一种类似ALDH2抑制剂的物质,和特殊的ALDH2抑制剂——黄豆苷,可通过纯化的ALDH2抑制1,2-GDN形成。还有,乙醛在体内和体外通过ALDH2可竞争性抑制GTN翻转。ALDH2失活涉及的机制可能是包括活性部位半胱胺酸硫醇二硫键形成的结果(图1)。如前所述,ALDH2的催化部位含有3个毗邻的硫氢基团,能形成分子内部的二硫键,导致ALDH2的失活。
活性部位二硫化物形成是ALDH2受GTN、NO、它的底物如4-羟基壬烯以及其它ALDH抑制剂抑制的基础。在体外对纯化的ALDH2分析表明,用外源性硫醇或其它还原剂对酶氧化的还原,可恢复ALDH2活性。线粒体ALDH2还成为GTN耐药超氧化物假说的集合点。超氧化物和过氧亚硝酸盐可直接抑制ALDH2活性。Daiber等观察到GTN-刺激的超氧化物产生与ALDH2活性降低密切相关。而且,由线体呼吸阻塞介导的活性氧产生与线本ALDH2失活有关。Sydow等报道动物模型中,GTN耐药的进展与血管ALDH2活性的抑制、GTN代谢的破坏及线粒体活性氧产生增多直接相关。有趣的是,在培养的缺乏线粒体的内皮细胞中,对GTN发生反应的cGMP增加是迟钝的。这些所见支持这种假说,即活性氧的释放和反应性醛类如4-羟基-2-壬烯醛的进一步积累,通过ALDH2的氧化抑制或氧化酶辅因子,可直接引起GTN耐药。事实上,与其它有机硝酸盐的生物活性、或血管松弛信号下游成分相关的超氧化物诱导的关键酶抑制,可引起GTN介导的交叉耐药。有趣的是,耐药组织与不同减量药物或抗氧化剂分子孵育可改善血管ALDH2活性,因此支持这种假说,即酶氧化导致GTN耐药。最近研究表明,在试管内醛缩酶-1可轻度抑制GTN生物转化成NO,考虑到NO对心绞痛患者的医疗益处,这是令人不感兴趣的。然而,我们发现随GTN给予醛缩酶-1在体内并不抑制血管扩张,表明在体内醛缩酶-1不抑制GTN生物转化成NO。然而,我们发现,醛缩酶-1可防止长期用GTN治疗引起的ALDH2失活(图2)。
对心肌细胞的影响
在血管床内GTN的作用已得到了广泛的研究,但关于GTN对心肌细胞的影响所知较少。我们最近已经证明,在老鼠MI后用GTN持续治疗可引起梗死面积增加和心功能不全。GTN耐药介导的心脏恶化作用与ALDH2失活有关。正如以往所报道的,GTN治疗显著抑制重组ALDH2在体内外的脱氢酶活性。我们还发现,用GTN和醛缩酶-1共同孵育可完全预防GTN诱导的重组ALDH2失活。而且,用GTN持续治疗可显著降低老鼠心肌内的线本ALDH2活性,引起心肌梗死后的心肌损害增加和心室功能不全。有趣的是,用GTN和醛缩酶-1共同治疗可恢复ALDH2活性,可使老鼠的心梗面积减少、心脏功能改善。我们提出GTN耐药是主要的涉及心梗后心肌损害增加的过程,因为硝酸异山梨酯(常以维持方式用于治疗心绞痛的另一种NO供体)的使用,在体内不引起ALDH2失活和进一个步心肌损害。与此相似,Sydow等观察到,在体内用GTN治疗可因线粒体ALDH2导致心脏GTN生物转化降低,并引起活性氧的积累,而用取自耐药动脉的线粒体与减量的药物孵育可恢复ALDH2功能。这些所见提示用GTN连续治疗的患者处于心脏损害增加的风险。
GTN的心脏负作用
有机硝酸盐治疗对线粒体的有害作用首次于55年前被描述,当时描述急性GTN暴露可引起线粒体肿胀、刺激氧耗、并引起老鼠肝脏和心脏线粒体呼吸控制的丧失。较近证实在动物和人体血管GTN输注可引起线粒体功能不全诱导的氧化应激。对孤立的线粒体应用复合体III抑制剂抗酶素A,可见到过多的活性氧产生以及ALDH2活性降低50%以上。以线粒体为靶点的抗氧化剂可预防由GTN治疗介导的复合体I抑制。因此,来源于氧化应激的反应性乙醛积累,通过负面作用于ALDH2功能可破坏GTN生物活性。总之,这些所见提示GTN的生物活性需要功能上有活性的线粒体,因为由线粒体功能不全而增多的活性氧产生,可引起ALDH2活性和进一步的GTN转化受损。我们最近发现,持续用GTN治疗明显降低衰竭心脏的乙醛脱氢酶活性。哺乳动物的ALDK2功能作为GTN还原酶,这一事实可解释GTN对乙醛脱氢酶活性的抑制作用。这些所见表明通过ALDH2失活酒精-GTN可能存在相互反应,由于某种原因细胞内反应性乙醛的积累,可能引起灾难性现象。
ALDH*2突变与GTN代谢
40%以上的东亚人携带一种常见的ALDH2*2突变。突变是由于12号染色体上ALDH基因的12外显子被一个单核苷酸取代引起的,导致成熟酶487位点(E487)的谷氨酸转变为赖氨酸。在四聚体酶的接触面的E487氨基酸取代,因为辅酶NAD结合的破坏,导致ALDH2催化活性的降低。杂合子ALDH2*1/*2和纯合子ALDH*2/*2,分别显示出40%和5%的野生型ALDH2活性。
流行病学研究表明,在心血管疾病中ALDH是很重要的,ALDH*2/*2等位基因与较高的GTN耐药、MI和高血压发生率相关联。考虑到ALDH2对GTN生物活性的影响,预期在携带E487突变的亚洲人中,ALDH2活性显著降低可引起对GTN治疗的血管扩张反应降低。事实上,在试管内实验证实用E487突变酶GTN生物转化显著降低。携带ALDH*2/*2突变者对GTN的血管扩张反应明显降低,因此,需要更大剂量的GTN才能达到满意的血管扩张。有趣的是,非ALDH2突变个体用ALDH2抑制剂戒酒硫治疗,可阻滞GTN的血管扩张作用,但不被硝普钠所促进。这些所见证实,在人体内ALDH2对GTN的生物活性是很重要的,携带ALDH*2/*2突变的个体,表现出对GTN的血管扩张反应降低,并可能更容易发生与心脏毒性相关的GTN耐药。
临床意义和总结
ALDH2对GTN生物活性是很重要的,这一发现可能有重要的临床意义。考虑到ALDH2活性在缺血事件过程中与心脏保护正相关,降低ALDH2活性的(即持续用GTN)治疗,在缺血发作过程中可能使预后恶化。GTN是最常用的治疗心绞痛、缺血和心力衰竭的药物。然而,关于长期GTN治疗的恶化作用存在争论。考虑到ALDH2对不同组织中细胞保护信号的重要性,GTN耐药不应看作药效的简单丧失。对缺血事件风险增高的患者,应当评估当前GTN治疗的临床实践。持续暴露于GTN应当避免,不仅其益处会被消磨,也许更重要的是,还因为它降低ALDH2的活性并因此加重与缺血相关的损害。能够增强GTN生物转化和预防GTN抑制ALDH2的药物,将是很有用的,特别是对ALDH*2/*2个体。ALDH2激活剂例如醛缩酶-1,因为其能防止GTN引起的ALDH2失活,可能有潜在的治疗价值。醛缩酶-1能恢复突变ALDH*2/*2的活性,这一事实提示这些个体在用了醛缩酶-1后可能变得对GTN治疗更有效(图3)。最后,需要进一个步研究以澄清GTN的生物转化和GTN耐药的分子机制,以及对心血管病患者,尤其是对东亚患者,持续应用GTN治疗的临床研究进行重新评估。
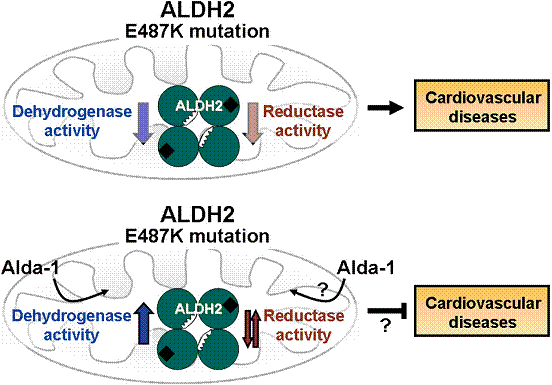
由487位(E487K)的单个氨基酸从鸟氨酸转换成赖氨酸引起的突变,可引起ALDH2脱氢酶和还原酶的活性降低。 醛缩酶-1明显改善突变酶的脱氢酶活性,引起酶变构改变,可纠正酶的结构缺陷。醛缩酶-1对ALDH2还原酶的作用目前还未明确。
译自
Julio C.B. Ferreira, PhD; Daria Mochly-Rosen, PhD:Circ J 2012; 76: 15–21
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#硝酸甘油#
41